
This symposium is the fifth in the series on Instrumental Analysis within the frame of the partnership between the cities of Pécs and Graz. The symposia were held in Pécs and in Graz alternatively since 1991 every second year. The scope of this symposium series is to give an overview of the activities in the field of instrumental analysis in both cities. It is expected that this conference will further contribute to the exchange of ideas and will provide a forum for stimulating discussions.
The symposium will be held at the Center of the Regional Committee of the Hungarian Academy of Sciences, Pécs, Hungary (Vasvári Villa, Jurisics M. u. 44., H-7617, Pécs).
Lunch is organized at the Faculty of Natural Sciences, Janus Pannonius
University
(Ifjúság útja 6.) The reception of the Mayor of
Pécs will be held at the Town Hall
(Széchenyi tér 1.)
Participants (foreign): 1200 ATS
Students (foreign): 600 ATS
Accompanying persons (foreign): 600 ATS
Participants (Hungarian) full price: 9500 HUF
Participants (Hungarian) reduced price: 4500 HUF
Registration fee includes participation, book of abstracts, lunches and the social program (welcome party, reception with dinner, excursion to the wine country and guided tour through the city) and accommodation in double room (single accommodation will be available with extra charge). Reduced price does not include lunches and the excursion
Posters should be mounted by 9:00 a.m., 25th October, and dismounted by 15:00 p.m., 26th October. Authors are requested to be present at their posters during the poster discussion.
19:00 Welcome reception
25th October, 1999
09:00 Opening Ceremony
Chairmen: Ferenc Kilár, Ernst Kenndler
09:10 L-1 Stellan Hjertén, Christer Ericson, Ákos
Végvári, Audrius Maruka, Olga Kornysova, Vilma Kudirkaité,
Egidijus Machtejevas
Electrochromatography on continuous beds
10:00 L-2 Martin G. Schmid, Gerald Gübitz,
Chiral separation of various compound classes by ligand exchange
capillary electrophoresis
10:20 L-3 Ákos Végvári, Christer Ericson,
Stellan Hjertén
Advantages of homogeneous gels in CEC applications
10:40 Coffee break
Chairmen: Stellan Hjertén, Gerald Gübitz
11:00 L-4 Ernst Kenndler
Capillary electrophoresis of viruses and subviral particles
11:40 L-5 T. Wenzl, E. Lankmayr
Migration studies of packaging material
12:00 L-6 Gabriel Peltre
Allergens studied by the Electro Titration Curve technique.
12:20 Lunch
-----------------------------------------------------------------
13:30 Poster discussion
P-1 S. Trummer , P. Sedlmayr, K. Wascher, A. Hammer, A. Blaschitz,
W. Walcher, R. Wintersteiger, G. Dohr
Absence of HLA-G protein on macrophages in first trimester decidua
P-2 S. Strassnig, E.P. Lankmayr
Optimization of a fast derivatization procedure for volatile carbonyl
compounds after trapping on TENAX
P-3 F. Stelzer, R. Bona, G. Braunegg
Analysis of polyalcanoate copolymers by means of pyrolysis gas chromatography
P-4 G. Schwarzenbacher, C. Mirtl, M.S. Gangl, M. Goriup, R.
Saf
End group modification of poly(ethylene glycol)s: electrospray ionisation
mass spectrometry as analytical tool for subsequent optimisation of reaction
conditions
P-5 C. Schür, M. Leicht, T. Marek, H.P. Strunk
Convergent beam electron diffraction for the determination of the
tetragonal distortion of cubic crystals, demonstrated on "low temperature
grown" GaAs
P-6 Balázs Schäffer, Dénes Lõrinczy,
Béla Schäffer
Thermodynamic characteristics of dispersion-type processed cheeses
made without peptization by DSC methods
P-7 K. Rittstieg, W. Somitsch , K.-H. Robra
Quantification of the bleaching agent "FAS and its decomposition
products by HPLC
P-8 Harald Reisinger, Karin Viertler, Alf Wewerka, Peter Preishuber-Pflügl,
Franz Stelzer
Homopolymers, statistical copolymers and block-polymers of norbornene
derivatives: characterisation by means of chromatographic methods
P-9 Zoltán Pintér, Tamás Lóránd
GC-MS investigation of isomeric E- and Z-4-arylidene-3-isochromanones
P-10 Andrea Petz, Zoltán Tuba, Zoltán Pintér,
Zoltán Berente, László Kollár
Synthesis and characterization of steroidal crown ethers
P-11 K. Lübke, E. Leitner, W. Pfannhauser
SPME - a simple sample preparation method for migration studies
P-12 H.J. Leis, G. Raspotnig, W. Windischhofer, G. Fauler
Determination of estriol in human plasma by stable isotope dilution
gas chromatography-negative ion chemical ionization mass spectrometry.
P-13 O. Lecnik, Martin G. Schmid, Gerald Gübitz
Enantioseparation of beta-blockers, sympathomimetics and hydroxy
acids by ligand exchange capillary electrophoresis
P-14 Wolfgang Kern, Ursula Meyer, Thomas Kavc, Eva-Maria Hoiser,
Maria F. Ebel, Robert Svagera , Peter Pölt
Surface characterization of photochemically modified polymers
P-15 Susan Juricskay
Combined TLC and capillary gas chromatographic method for the quantification
of phytanic acid and very long chain fatty acids in serum of patients with
peroxisomal deficiencies
P-16 Milan Hutta, Mária Chalányová, Róbert
Góra, Radoslav Halko, Marek Pajchl, A. S. Dokupilová
Development of RP-HPLC method for determination of selected group
of pesticides in soils
P-17 Róbert Góra, Milan Hutta, Ján Kandrács
Characterization of soil humic substances by liquid chromatography
methods
P-18 Marion Gfrerer, Thomas Wenzl, Ernst Lankmayr
Method development for broad range screening and simultaneous determination
of biocides in river samples from Eastern China
P-19 T. Boyko ,W. Kosmus
Metal associations in carious teeth of industrial areas of Ukraine
P-20 Ildikó Kerepesi, Éva Stefanovits-Bányai,
Zoltán Szabó, Éva Sárdi, István Pais
Influence of Ti(IV)ascorbate on carbohydrate metabolism in cadmium
treated wheat
P-21 Klára Szabó, K. Szaláncz, Nándor
Marek
Study on a catalytic chemiluminescence reaction
P-22 A. Loidl, R. Claus, P. Deigner, A. Hermetter
A fluoresence sphingomyelinase assay using a CCD camera
P-23 Stephan Landgraf
Application of semiconductor light sources for time-resolved fluorescence
measurements.
25th October, 1999
P-24 H. Brandstätter, B. Mayer, G. Hofer, A. Hermetter
Fluorescence monitoring of free radical-mediated lipid oxidation
and its inhibition by antioxidants in serum
P-25 J. Deli , P. Molnár , E. Õsz , G. Tóth
, A. Haberli, H. Pfander
Isolation and characterisation of carotenoids from the fruits of
Asparagus falcatus
P-26 György Petõcz , László Jánosi,
Zoltán Berente, László Kollár
Synthesis and NMR investigation of Pt(CN)2(diphosphine)
and [Pt(CN)(triphosphine)]Cl complexes
P-27 Pál Perjési, Alexander Perjessy
Investigation of transmission of substituent effects in E-2-benzylidene
derivatives of cyclohexanone and tetralone by IR spectroscopy and theoretical
methods
P-28 Tamás Lóránd, Sándor Holly,
Gábor Keresztury
FT-IR and Raman study of some new E- and Z-4-arylidene-3-isochromanones
P-29 Tamás Lóránd, József Deli,
Péter Molnár, Gyula Tóth
FT-IR study of some carotenoids
P-30 Barna Kovács, Roland Dombi , Géza Nagy
Flow injection detection of urea using a planar waveguide biosensor
as detector
P-31 Márta Kiss, Nóra Hartvig, Dénes
Lõrincy, József Belágyi
Effect of oxygen free radicals on the internal dynamics of actin
P-32 Nóra Hartvig, Balázs Gaszner, Balázs
Schäffer, Dénes Lõrinczy, József Belágyi
Global and local conformational changes in myosin induced by AMP.PNP
P-33 Kornélia Farkas, Miklós Pesti, József
Belágyi
EPR study on chromium-sensitive and resistant mutants of Schizosaccharomyces
pombe
P-34 Balázs Csóka, Barna Kovács , Géza
Nagy
Determination of ionic surfactants by solid state potentiometric
sensor array
-----------------------------------------------------------------------
14:45 Coffee break
Chairmen: Reinhold Wintersteiger, Franz Stelzer
15:00 L-7 P. Sedlmayr
Principle and applications of flow cytometry
15:40 L-8 Sándor Kunsági-Máté
Spectrofluorometric and quantum-chemical studieson host-guest interaction
of calixarene and benzotrifluoride derivatives
16:00 L-9 H. Holzer
Akkreditierung von Chemischen Prüflabors und Validierung von
Analysenverfahren
16:30 L-10 Pál Perjési, Pál Sohár,
Richard E. Bozak
Synthesis and stereochemistry of some cyclic chalcone derivatives
and their ferrocene analogues
19:00 Reception by the Mayor of Pécs
26th October, 1999
Chairmen: Walter Kosmus, Wolfgang Kern
09:00 L-11 Géza Nagy, Klára Tóth, Ernõ
Lindner, Michael R. Newman, Alessandra Christalli, Robert Gyucsányi
Recent results with enzyme activity measuring biosensors
09:40 L-12 H. Greschonig
The electroanalytical determination ofinorganic and organic arsenic
compounds
10:00 L-13 Imre Sánta
Femtochemistry
10:20 L-14 J. Erostyák, A., Buzády B., Somogyi
Time-resolved spectroscopy measurements by phase-fluorometer
10:40 Coffee break
Chairmen: Ernst Lankmayr, Hans Jörg Leis
11:00 L-15 W. Gruber, W. Trettnak, V. Ribitsch
Luminescence based sensors - Basic principles and applications
11:40 L-16 B. Goger, R. Wintersteiger
Quantification of pesticides by HPLC/ECD and photolysis
12:00 L-17 Miklós Nyitrai, Gábor Hild, András
Lukács, Emõke Bódis, Szulamit Halasi And Béla
Somogyi
The dynamic and conformational properties of the catalytic and light-chain-binding
domains of s1 in the acto-myosin complex
12:20 L-18 Zoltán Berente, Erzsébet Õsz,
Balázs Sümegi
NMR studies of metabolic processes
15:00 Excursion to the wine country
27th October, 1999
09:30 Guided tour in Pécs
12:00 Lunch
ELECTROCHROMATOGRAPHY ON CONTINUOUS BEDS
Stellan Hjertén*, Christer Ericson, Ákos Végvári, Audrius Maruka, Olga Kornysova, Vilma Kudirkaité, Egidijus Machtejevas
Department of Biochemistry, Biomedical Center, Uppsala University, Box 576, Uppsala, SE-751 23, Sweden
In capillary electrochromatography (CEC) the mobile phase is propelled through the stationary phase by electroendosmosis (no mechanical pump is thus required), i.e., the bed must be derivatized with charged groups. As in all chromatographic experiments the separations are based on interactions between the analytes and the bed or the attached ligands, for instance C18 groups.
The continuous beds can with advantage be used in CEC since they have the following advantages over beds packed with beads.
L-2
CHIRAL SEPARATION OF VARIOUS COMPOUND CLASSES BY LIGAND EXCHANGE CAPILLARY ELECTROPHORESIS
Martin G. Schmid and Gerald Gübitz
Institute of Pharmaceutical Chemistry, Karl-Franzens-University Graz, Universitätsplatz 1, A-8010 Graz, Austria
Ligand exchange capillary electrophoresis has successfully been applied to the direct resolution of underivatized amino acids using L-proline, L-4-hydroxyproline and N-alkyl-L-4-hydroxyproline in form of their copper(II) complexes as chiral selectors [1, 2]. This contribution deals with the application of this basic approach for the enantiomeric separation of hydroxy acids, dipeptides and drugs containing amino alcohol structure.
Copper(II) complexes of N-alkyl-L-4-hydroxyproline such as N-propyl- and N-octyl were compared for their ability of resolving amino alcohols and hydroxy acids.
In addition to amino acids, hydroxy acids, dipeptides and several drugs containing amino alcohol structure i. g. sympathomimetics and ß blockers were resolved into their enantiomers. Hydrophobic interactions of the lipophilic chain of the selector and the alkyl groups of the analytes might enhance selectivity. The influence of the concentration of the chiral selector, the copper(II) content, the electrolyte composition and the pH as well as the addition of organic modifiers on the enantioselectivity was investigated. For the separation of amino- and hydroxy acids the pH optimum was found to be 4.3, whereas dipeptides were optimally resolved at pH 6 and amino alcohols at pH 12. Since in HPLC it is not possible to work at high pH, CE is advantageous for the chiral separation of this compound class using the principle of ligand exchange.
[1] M.G. Schmid and G. Gübitz, Enantiomer, 1, 23-27 (1995)
[2] Á. Végvári, M.G. Schmid, F. Kilàr and G. Gübitz, Electrophoresis,19, 2109-2112 (1998)
L-3
ADVANTAGES OF HOMOGENEOUS GELS IN CEC APPLICATIONS
Ákos Végvári1, Christer Ericson2, Stellan Hjertén2
1Central Research Laboratory, Medical University of Pécs, Szigeti út 12, Pécs, H-7643, Hungary
2Department of Biochemistry, Biomedical Center, Uppsala University, Box 576, Uppsala, SE-751, 23 Sweden
Completely homogeneous gels have pore sizes which are much smaller than the average diameter of the channels between the beads in conventional packed beds. Accordingly, in CEC the residence time of the analytes in the mobile phase is shorter (the third term in the van Deemter equation). The gels have the great advantage to generate a perfect electroendosmotic plug flow and, therefore, the Eddy diffusion is negligible (the first term in the equation). The longitudinal diffusion is restricted (the second term). In terms of the van Deemter equation the resolution is, accordingly, high. The preparation of these gels is simple. The bed is attached to the wall and no frit is required to support it.
These attractive electrochromatographic features will be demonstrated, using different matrices (cross-linked polyacrylamide, polyvinylalcohol and agarose). Separations of nucleosides, enantiomers and alkyl esters of p-hydrobenzoic acids will be presented utilizing different types of interaction.
L-4
CAPILLARY ELECTROPHORESIS OF VIRUSES AND SUBVIRAL PARTICLES
Ernst Kenndler
Department of Analytical Chemistry, University of Vienna, Währingerstr. 38., Vienna, A 1090 Austria
Nm-particles possess an electrically charged surface in most cases, and consequently form an electrical double layer, characterised by the zeta potential. Therefore they migrate in an electric field with mobility according to the theory of Hückel, Smoluchowsky and Henry. Even colloidal particles of elementary gold exhibit this behaviour, their surface charge stemming from ion adsorption in this case.
Viruses, particles with nm size, too, consist of a protein capsid, which encloses the genom. It seems obvious that the protein shell exhibits a surface charge as well, giving raise to migration upon application of an electrical field. The electrophoretic behaviour of virions is expected to be modified upon binding of antibodies, soluble virus receptors or small antivirally active compounds, because such interactions can lead to variations of the surface charge, changes in mass, modification of the virus conformation, or to a combination of these effects. Capillary electrophoresis is assumed to be a favourable technique to investigate the interactions of viruses with neutralising agents because it allows to apply physiological conditions by selecting the appropriate (homogenous solution) of the background electrolyte.
Different modes of CE enable to analyse various molecular forms of conformational states of viruses (human rhinovirus HRV2). Capillary isoelectric focusing of HRV2 enables to determine rapidly the isoelectric point of the intact virus on the one hand, and of the protein capsid resulting from denaturation of the virus on the other hand. However, the accurate identification of the peaks occurring in the electropherograms is not a trivial task. Two approaches to support confirmation are discussed:
MIGRATION STUDIES OF PACKAGING MATERIAL
T. Wenzl, E. Lankmayr
Institute of Analytical Chemistry, Radio- and Mikrochemistry
Technical Univeristy of Graz, Technikerstr. 4, A-8010 Graz, Austria
The main purpose of packaging materials is to preserve the quality of packaged food, which may be affected by light, oxygen, micro-organisms and the uptake or release of water. Beside this, there are regulations to preserve the consumer from negative influences of the packaging onto the consumers health, originating from mechanical and thermal treatment or from chemical composition.
An important point of view, which is the topic of this work and which is also demanded in the regulations for food packaging, is robustness against migration. In guideline 76/893 of the EWG (now European Union), it was already specified that "there should not be a transference of substances from the packaging into the food , which could endanger humans health.
Migration may occur through the gas phase as well as by direct contact between the package and the food. Well investigated are the migration properties of substances in plastic packages, like residual monomers, dyes and especially plasticisers, due to their high concentration. However, also cellulose based packaging material may contain migrable components. Special emphasis has to be given to substances which change the sensorial characteristics of the product. This was the background for the investigation of volatile aldehydes in recycling cardboard.
The most important parameters in migration are diffusion and partition. Whereas the diffusion mainly depends on temperature and time, for a given system, partition is predominantly determined by temperature and the solubility of the migrating substance in the adjacent phase. Systematic studies encounter the problem of inhomogeneous and temporarily varying matrices.
Further problems originate from differences of the chemical properties of the investigated matrices. The fat and water content of a product e.g. have a significant impact onto distribution characteristics. Therefore constant matrix compositions, like distilled water, acetic acid, ethanol solutions in water, olive oil are synthetic fats, are commonly used to obtain comparable results of analysis [2]. The disadvantages related to these systems are obvious. They are liquid and difficult to separate from the matrix, when they are brought into direct contact.
Therefore special attention was given to an investigation of the migration through the gas phase as well as by direct contact. Further research was done on the suitability of Tenax TA as food simulating agent. Official regulation include an analysis parameter, which is called global migration. By extraction of the packaging material with an suitable solvent, the quantity of potential migrating substances is determined. A new commercial available extraction system was tested for this purpose.
[1] Buchner, R. Verpackung von Lebensmitteln, Springer-Verlag Berlin Heidelberg, 1999
[2] Guidline of the European Union: 97/48/EG
L-6
ALLERGENS STUDIED BY THE ELECTRO TITRATION CURVE TECHNIQUE.
Gabriel Peltre
Immuno AllergyUnit, Institut Pasteur, 28 rue du Dr. Roux, 75724 (15) Paris, France
The Electro Titration Curve (ETC) technique is an excellent analytical method, if not the best, which gives the isoelectric points of each molecule present in an heterogenous sample. It gives also the electrophoretic mobility of each molecule in the pH range tested. This added information can be very precious for any further preparative electrophoretic or chromatographic purification, based on the molecular charge, of the different components present in this sample. The ETC technique has furthermore the advantage to be a very mild technique that does not modify strongly the protein structure as it is performed quickly (15 minutes) in a 2% ampholyte solution in water supported in a porous gel, at a non denaturing temperature.
The ETC allows an easy and fast blotting, especially if it is performed in a soft agarose gel. The allergen identification is then done by detecting allergic patients' IgE or allergen specific monoclonal antibody binding.We have observed, in a limited pH range, some interactions between several purified major allergens from grass pollen extracts. These interactions, evidenced in the allergenic extracts up to now only between known allergens, may be related to the very special role of this family of molecules that makes them inducing and being recognized by IgE molecules.
These properties of the ETC should be exploited to study other categoties of sensitive molecules such as enzymes, hormones, receptors, cytokines, lectines whose function is greatly dependent upon a"native" structure or conformation
L-7
PRINCIPLE AND APPLICATIONS OF FLOW CYTOMETRY
P. Sedlmayr
Institute for Histology and Embryology , Karl-Franzens-University, Harrachgasse 21, A-8010 Graz, Austria
Flow cytometry (FCM) is a technique well established in immunology and cell biology. It enables us to measure many on the surface and inside cells in a qualitative and quantitative manner. FCM combines hydrodynamic focusing with the measurement of light scattering and fluorescence. Typically, cells in a liquid stream pass the sensing area in a flow cell one by one. The interception of a laser beam with these cells results in scattering of light. The scatter is measured by a photodiode at near 180° (forward scatter, FCS), but also by a photomultiplier at 90° (side scatter, SSC). FSC correlates to cell size, SSC to granularity and roundness of the nucleus. Further parameters are added due to the fluorescence of the cells which is measured by photomultipliers after passing a set of optical filters and dichroitic mirrors.
In addition to simply measuring cells at speeds up to several thousands per second and correlating all the measured parameter to each single event, the cells can be physically separated and collected ("sorted") in some instruments. These devices use piezoelectric quartz crystals of which the oscillation is transduced to a liquid jet in air, leading to a very stable delay of droplet formation. At the time when a droplet forms which contains a cell previously defined as being inside a region of interest, a voltage pulse is applied to the jet. So the droplet containing the cell of interest is electrically charged and can be deflected into a separate vial using an electrostatic field.
There is some low level of background fluorescence due e. g. to the presence of naturally occurring coenzymes. Fluorescence becomes much brighter after labeling certain molecules with fluorochromes. There are e. g. dyes binding specifically to DNA, enabling a cell cycle analysis, as cell in G2 phase contain twice the amount of DNA as cells in Go/G1 phase, the S phase cells being in between. FCM also makes extensive use of the principle of immunofluorescence, where labeled antibodies bind to specific molecules on the cell surface or - following cell membrane permeabilization - also in the cytoplasm. Further examples of a wealth of applications of FCM include measurement of RNA or total cell protein, of intracellular pH or Ca++, of the membrane potential of cells or organelles, assessment of cell proliferation, phagocytosis and respiratory burst, membran fluidity and enzymatic activity.
L-8
SPECTROFLUOROMETRIC AND QUANTUM-CHEMICAL STUDIES ON HOST-GUEST INTERACTION OF CALIXARENE AND BENZOTRIFLUORIDE DERIVATIVES
Sándor Kunsági-Máté
Department of General and Physical Chemistry, Janus Pannonius University, Ifjúság 6., H-7601 Pécs, Hungary, E-mail : kunsagi@ttk.jpte.hu
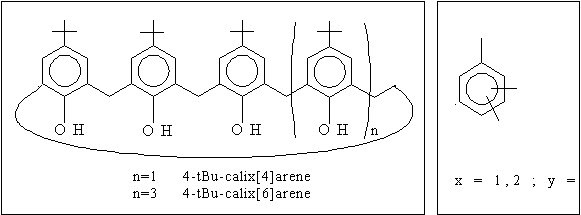
Calixarene derivatives have photoluminescent behaviour at near ultraviolet range. This characteristic property shows significant changes when the molecule interacts with ions. Different calixarene derivatives have been successfully used for photoluminescent imaging of different ions in histology. Some conformers (cone, partial cone) of these derivatives proved to be excellent host molecules in their interaction with organic guests which makes calixarenes promising candidates for chemical sensors.
Recently we studied the 'host' properties of calixarene compounds (Fig.1.) under the effect of uncharged benzotrifluoride (BTF) molecules (Fig.2.). The samples (mixtures of calixarene and BTF derivatives) were prepared in chloroform and dimethylformamide solvents with equimolar concentration (ccalixarene = cBTF = 10-4 mol/dm3 ). The photoluminescence spectra of these samples show a dependence on the type of BTF derivative. We interpret this dependence as a second-ordered interaction between calixarene and BTF molecules.
The photoluminescence spectra were measured on Fluorolog-3 /Jobin-Yvon-SPEX/ spectrofluorometer. In order to model the interaction of calixarene and BTF derivatives quantum-chemical methods were used. The interaction energy curve was determined by the total energy (semi-empirical and Hartree-Fock level, followed by semi-direct second order Moller-Plesset perturbation treatment). The conformations were optimized for minimum energy.
The results have clearly indicated the possible formation of stable complexes of calixarene and some BTF derivatives.
L-9
Bundesanstalt für Lebensmitteluntersuchung Graz
Die staatlichen Prüflabors im Lebensmittelbereich sind akkreditiert. Die Normen EN45001 und EN45004 sind die Basisnormen für amtliche chemische Prüflabors zu deren erforder-licher Akkreditierung.
EAL-Dokumente liefern Interpretationen und Anleitungen zur Umsetzung von Elementen eines Qualitätssystems.
Das Qualitätssicherungshandbuch (QSHB) bildet neben weiteren wichtigen mitgeltenden Unterlagen das dokumentarische Kernstück des Qualitätssystems. Darin müssen u.a. die Aussagen zur Qualitätspolitik der Prüfeinrichtung präzisiert sein.
Weitere wesentliche Komponenten sind die Standardarbeitsanweisungen (SOP). Dabei handelt es sich um die Prüfvorschriften und um solche Arbeits- bzw. Verfahrens-anweisungen, die nicht in den Prüfvorschriften enthalten sind. Für bestimmte Elemente des Qualitätssystems (außer den Prüfvorschriften) sind SOPs verpflichtend vorge-schrieben.
Zur Überwachung der Prüf- und Messmittel besteht für jedes Gerät ein Gerätehandbuch, in dem nachvollziehbar die lückenlose Dokumentation des laufenden Zustandes, der diesbezüglichen Überprüfungen, die erfolgswirksamen Maßnahmen und ggf. die Sperre des Gerätes protokolliert sind.
Begriffe wie Validierung, Zertifizierung, Charakterisierung, Verifizierung und Qualifizierung geben unter Umständen Anlass zu Verwirrung. Vom Begriff Validierung bestehen in der Literatur mehrfach Definitionen. Ein (weiterer) Normierungsentwurf liegt vor.
Die in der Routine eingesetzten Prüfverfahren müssen validiert sein. Die Validierungs-parameter (Analysenkenndaten) müssen in den Prüfvorschriften enthalten sein.
Die Befassung mit Normen gehört zum Arbeitsalltag, weil von vielen Begriffen ein-schließlich der Analytik (trendgemäß) Normen bestehen, relativ kurzfristig geändert oder neu verfasst werden.
Für konkrete Anwendungssituationen erweisen sich manche Normierungen als nicht wesentlich hilfreich.
Durch interne Audits und schließlich jährlich zu erstellende Review-Berichte mit der Konsequenz der Nachbesserung oder der Einführung von Verbesserungen bleibt das Qualitätssystem dynamisch bzw. vital.
L-10
SYNTHESIS AND STEREOCHEMISTRY OF SOME CYCLIC CHALCONE DERIVATIVES AND THEIR FERROCENE ANALOGUES
Pál Perjésia, Pál Sohárb, Richard E. Bozakc
aInstitute of Medical Chemistry, University Medical School, Pécs, Hungary
bDepartment of General and Inorganic Chemistry, Eötvös Loránd University, Budapest, Hungary
cDepartment of Chemistry, California State University, Hayward, USA
Numerous synthetic and natural alpha,beta-unsaturated carbonyl compounds exhibit remarkable biological activity. Among others, several chalcones and related benzylideneketones have been reported to possess antifungal, antibacterial and cytotoxic (antitumor) activity.
Recently we have synthesized some cyclic chalcone derivatives such as E-2-benzylideneindanones (1a), -tetralones (1b), -benzosuberones (1c) and their ferrocene analogues (2a-c). We have investigated their in vitro antifungal [1,2] and anti-leukemia cytotoxic [3] activities. The observed biological activities have been found to be affected by both the ring size and the substitution pattern of the benzylidene moiety.
The biological activity of the compounds is considered to be associated with their reactivity with the thiol moiety of the critical peptides and/or proteins. Is this assumption true, then electronic and steric factors should affect the reactivity, i.e. their biological activity. In order to investigate the electronic and steric structures of the compounds we have performed a comparative IR, 1H and 13C NMR study on some selected derivatives.
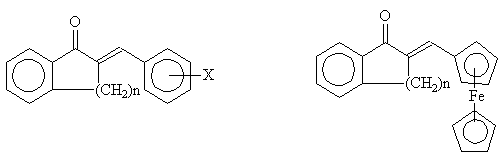
1 2
n=1(a), n=2(b), n=3(c)
X= H, OCH3, F, NO2
References:
[1] T. M. Al-Nakib, P. Perjési, R. Varghese, M. J. Meegan: Benzylideneindanones and benzylidenebenzosuberones: Relationship between structure, antimycotic activity and acute toxicity.
Med. Princip. Pract. 6, 14-21 (1997).
[2] P. Perjési, T. M. Al-Nakib, M. Abdulhamid: Structure-activity relationship and acute toxicity of new alpha,beta-unsaturated ketones
Health Sciences Poster Day, Faculty of Medicine, Kuwait University (Kuwait), April 20, 1998.
[3] R.E. Bozak, R. J. Hicks, P. Perjési: The anti-leukemia cytotoxicities of some ferrocenyl enones and their phenyl analogues.
Pacific Conference of the American Chemical Society, San Francisco (USA), October 28-31, 1998.
L-11
RECENT RESULTS WITH ENZYME ACTIVITY MEASURING BIOSENSORS
Géza Nagy1, Klára Tóth2, Ernõ Lindner3, Michael R. Newman3, Alessandra Christalli3, Robert Gyucsányi2
Janus Pannonius University, Department of General and Physical Chemistry, Pecs, Hungary
Technical university of Budapest, Institute for General and Analytical Chemistry, Budapest, Hungary
University of Memphis Department of Biomedical Engineering, Herff College of Engineering, Memphis TN, USA
Enzyme activity measurements are routinely carried out in different areas of analytical chemistry. As it is well known, the activity values of numerous enzymes measured in different clinical samples are considered in making clinical diagnosis. The practice of enzyme labeled immune assay rely on the measurement of the activity of the labeling enzyme. Further more sensitive analytical rate methods applicable for environmental samples are based on the enzyme action inhibiting effect of the analyte.
Enzyme activity measuring probes have already been prepared in the early times of biosensor research. However, their development, and application obtained little attention and mostly conventional spectroscopic methods have been dominating in the enzyme activity determinations. It was supposed that sensitive electrochemical detection combined with specific cell design could be advantageously used to prepare enzyme activity measuring biosensors and to work out biosensor based procedures.
In our work to be presented a novel flat form amperometric measuring cell type and a potentiometric biosensor were investigated to develop analytical procedures based on enzyme activity detection.
The amperometric working electrodes detect the concentration of an electroactive species in the liquid layer adjacent to their concentration. Therefore a thin layer type cell with very low sample volume requirement can be used very effectively in combination with them. Kapton based flat form cells made with thin layer technology and alumina substrate based flat form cells with screen printed electrodes were prepared and tested with different enzyme analytes. In case of small volume cells holding just a few microliter solution the temporary isolation of the reference electrode can easily happen. In three-electrode system it can result in excessive current intensity depending on the limiting voltage value of the potentiostat used. This current shock can damage the working electrode. To avoid this happening, two-electrode cell construction was used.
In every cell chloride coated silver plate served for reference while the working electrodes were made of platinum or graphite. The platinum working elecrodes were elecrolytically coated with size exclusion membrane and they detected the hydrogen peroxide trough its amperometric oxidation. Oxidoreductase enzymes such as putrescine oxidase (EC 1.4.3.10), glucose oxidase (EC 1.1.3.4) catalyzing H2O2 generating reactions can be directly measured with the amperometric cell while in case of other enzymes like creatine kinase (EC 2.7.3.2) or (-amylase (EC 3.2.1.1) additional follow up reaction steps resulting H2O2 production were used. Experiments were carried out to work out dry reagent compositions adsorbed on porous material for the simplification of amperometric enzyme activity measurements with the platinum electrode holding flat form cell.
Two different enzymes of clinical importance were used to study the applicability of the planar cell equipped with graphite electrode in enzyme analysis, proline imino-peptidase (EC 3.4.11.5) and alkaline phosphatase (EC 3.1.3.1). The latter one, as a labeling enzyme is broadly used in ELISA methods too. Different substrates were used and the working conditions were optimized trying to minimize the fouling of the working electrodes.
In the other part of the work pH sensor based acethyl choline biosensors were prepared with a special procedure and its response was investigated after incubating it with enzyme inhibiting environmental pollutants. Procedure was worked out for the regeneration of the (choline esterase, EC 3.1.1.7) enzyme layer and in this way the potentiometric probe with enzyme activity dependent function could be repeatedly used. Owing to the separate incubation, and measuring step its pollutant measuring response was not affected by neither the pH nor the buffer capacity of the sample.
L-12
THE ELECTROANALYTICAL DETERMINATION OF INORGANIC AND ORGANIC ARSENIC COMPOUNDS
H. Greschonig
University of Graz, Austria, Institute of Analytical Chemistry,Universitaetsplatz 1
Arsenic is known as a toxic element and enjoys in a great extent the attention of scientists. Arsenic compounds are ubiquitous in the environment. It is the main constituent in more than 200 minerals and is also present in traces in a great variety of other minerals. Organisms are able to convert inorganic arsenic compounds into (methylated) organic species. Other industrial produced organic arsenic compounds were used as warfare agents, as pharmaceuticals or food additives and can also be found in the environment.
For a long time, the determination of the total arsenic concentration was the only measure for contamination. However, the very different toxicity of arsenic compounds promoted the necessity to develop methods for the identification and detection of arsenic species.
The first serious applicable method for the determination of total arsenic was the Marsh Test introduced in the middle of the 19th Century. Later, spectrometric methods, followed by the sophisticated inductively coupled plasma mass spectrometry became standard techniques for routine analysis.
Electroanalytical techniques were early used for the determination of inorganic arsenite and arsenate. Polarographic methods allow the determination of arsenite and arsenate, preferably in water samples, in the low ppm range. Under certain condition simultaneous determinations of the two inorganic species are also possible.
The low solubility of elemental arsenic in mercury prevents a successful stripping step during analysis. The addition of copper or selenium compounds supports the formation of mercury soluble arsenides. Thus, the detection limit for arsenite could be lowered to concentrations of approximately 500 ppb. The replacement of mercury by gold as electrode material lead to a further reduction of the detection limit towards approximately 40 ppb.
Organic arsenic compounds (besides electroinactive species containing four arsenic-carbon bonds) are also reducible under polarographic conditions. Selected pH values of background electrolytes allow the simultaneous determination of aliphatic and aromatic arsinic and arsonic acids regrettable at relatively high concentrations (about 10 mg /L). Substituents at the arseno group cause small shifts of reduction potentials. Therefore accurat identification of very similar species is not possible. Organic arsenic (III) and arsenic(V) compounds show very different chemical and toxicolgical properties. An important advantage of electroanalytical methods compared to other techniques is the possibility to identify different charged compounds. The irritant diphenylchloroarsine, for instance can be determined simultaneously with its less harmful oxidation product, diphenylarsinic acid.
Chemically modified carbon paste electrodes were also tested for their applicability in arsenic specification. They offer good sensitivity and selectivity, but the more complicated handling prevented a widespread practical use.
Capillary electrophoresis as a newer electrochemical separation technique offers excellent separations of a variety of charged and uncharged organic and inorganic arsenic compounds.
Gaseous arsenic compounds can be determined with a new type of amperometric gas sensor. Naturrally occurring arsines as well as chemically (by reduction with borohydride) synthesized arsines are oxidized at a gold or platinum coated Nafion foil. Speciation of arsenic compounds can be performed by using a simple chromatographic column as separation system.
L-13
FEMTOCHEMISTRY
Imre Sánta
(Abstract to be distributed later)
L-14
TIME-RESOLVED SPECTROSCOPY MEASUREMENTS BY PHASE-FLUOROMETER
J. Erostyáka,b, A. Buzádya,c, B. Somogyic
a Department of Experimental Physics, Institute of Physics, Janus Pannonius University
H-7624 Pécs, Ifjúság u. 6., Hungary (e-mail: erostyak@fizika.jpte.hu)
b Jobin-Yvon Reference Laboratory for Molecular Luminescence, Faculty of Natural Sciences, Janus Pannonius University
H-7624 Pécs, Ifjúság u. 6., Hungary
c Institute of Biophysics, Medical University of Pécs
H-7624 Pécs, Szigeti u. 12., Hungary
Time-resolved measurements are widely used in the fields of fluorescence spectroscopy. The basics and the main advantages of phase-fluorometry are shown first.
The temperature and viscosity dependence of fluorescence emission kinetics of tryptophan of Human Serum Albumin (HSA) was studied by the method of phase-fluorometry. Strong dependence of average fluorescence lifetime on the emission wavelength was found. The time-dependent spectral shift of fluorescence is characterized by the mean of fitting the phase-fluorometry data by a series of exponentials. The possible mechanisms of spectral shift is discussed analyzing the dependence of the mean emission energy, the solvation correlation function, the spectral width, the average lifetimes and the apparent lifetime components on the solvent viscosity and temperature. The influences of the experimental circumstances (purifying methods, buffers, instrument parameters, etc.) on the spectroscopy data are also discussed.
Acknowledgments. This work has been supported by the National Committee of Technical Development of Hungary under contract number 97-20-MU-0086.
References
[1] J. R. Alcala, E. Gratton and F. G. Prendergast, Biophys. J., (1987), 51, 925-936
[2] N. Rosato, E. Gratton, G. Mei and A. Finazzi-Agro, Biophys. J., (1990), 58, 817-822
[3] A. P. Demchenko, J. Gallay, M. Vincent and H. J. Apell, Biophys. Chem. (1998), 72, 265-283
[4] E. Laitinen, K. Salonen and T. Harju, J. Chem. Phys. (1996), 105, 9771-9780
[5] M. Vincent, J. Gallay and A. P. Demchenko, J. Fluorescence, (1997), 7, 107S-110S
[6] T. Fonseca and B. M. Ladányi, J. Phys. Chem. (1991), 95, 2116-2119
[7] X. Song, D. Chandler and R. A. Marcus, J. Phys. Chem. (1996), 100, 11954-11959
[8] R. Richert and M. Richert, Phys. Rev. E (1998), 58, 779-784
[9] J. R. Lakowicz, Topics in Fluorescence Spectroscopy, (1991), Plenum Press, N.Y.
L-15
LUMINESCENCE BASED SENSORS - BASIC PRINCIPLES AND APPLICATIONS
W. Gruber, W. Trettnak, V. Ribitsch
Institute of Chemical Process Development and Control, Joanneum Research, Steyrergasse 17, A-8010 Graz, Austria
Opto-chemical transducers are gaining increasing interest in chemical sensing technologies as they have certain advantages over existing transducers based on other physical-chemical effects. Among these, luminescence based transducers are well established in sciences but also are expanding to medical as well as industrial fields of research and development. Advances in optical technology now allow the practical application of techniques previously limited to research laboratories. For an increasing number of applications luminescence lifetime based sensing is the preferred method because of its inherent referencing possibility. Further, lifetime based instrumentation promises a simplified optical construction, since the measurement is, within certain limits, independent of the signal intensity.
Within the introduction, a discussion of the principles of methods for readout of the luminescence intensity and luminescence lifetime, being the output of an opto-chemical transducer, is given. Instrumentation schemes are compared with respect to information theory as well as practical aspects for the design of sensors. Theoretical limitations of various instrumentation technologies are addressed and practical examples are given. Especially for optical oxygen sensors, using dynamic fluorescence quenching as the information carrier, the advantages and disadvantages of lifetime based versus intensity based sensing are discussed. Finally, practical applications in the areas of biotechnology and industrial process control will be presented.
QUANTIFICATION OF PESTICIDES BY HPLC/ECD AND PHOTOLYSIS
B. Goger* and R. Wintersteiger
Institute of Pharmaceutical Chemistry, Karl-Franzens-University Graz,
Schubertstrasse 1, A-8010 Graz, Austria
Pesticides are toxic compounds which can be found in ground and drinking water because of their high solubility in aqueous medium. As the limits for pesticides in drinking water are set at 100ng/l1 for each individual compound, a highly selective and sensitive HPLC method in combination with electrochemical detection including photolysis was developed.
Different classes of pesticides like chlorophenoxy acetic acids2, phenylureas, benzimidazoles and some chlorinated compounds were investigated due to their electrochemical activity in the oxidative mode. Chromatographic conditions were optimized using a reversed-phase C18 column as stationary phase and methanol-water mixtures as mobile phase including acetic acid and lithium perchlorate as leading salt. Cyclic voltammograms and hydrodynamic voltammograms were recorded in order to determine optimum working potential. For the detection of the pesticides an amperometric and a coulometric detector in the range of +0.3V to 1.3V was applied.
As sufficient electrochemical activity is given at higher potentials, the influence of photolysis was studied for improving sensitivity and selectivity. Using UV-irradiation detection could be performed at potentials about 0.2V lower than without photolysis, thus improving selectivity. Some substances could only be detected under photolytic conditions. The extent of photoconversion processes is depending on the chemical structure.
The method developed was used for the limit determination of pesticides in Styrian drinking and ground waters. For efficient sample enrichment a C18 solid-phase extraction system has been developed resulting in recoveries between 80 and 100%. The majority of the compounds could be determined in the picomole range with good reproducibility. The method implies the option to be performed fully automated.
1 Economic Commission for Europe, Drinking Water Directive, 80/778/EEC, 1980
2 R. Wintersteiger and B. Goger, J. Chromatogr. A, 846 (1999) 349
L-17
THE DYNAMIC AND CONFORMATIONAL PROPERTIES OF THE CATALYTIC AND LIGHT-CHAIN-BINDING DOMAINS OF S1 IN THE ACTO-MYOSIN COMPLEX
Miklós Nyitrai1, Gábor Hild2, András Lukács1, Emõke Bódis2, Szulamit Halasi2 and Béla Somogyi1,2
1Research Group of the Hungarian Academy of Sciences at the 2Department of Biophysics, Medical University School of Pécs, P.O.B. 99, H-7601 Pécs, Hungary.
The combination of fluorescence resonance energy transfer spectroscopy and computer simulations provides the opportunity to determine the three-dimensional position of labeled points in actin-binding proteins relative to the actin filament. Using this method we have investigated the proximity relationship between the donor (IAEDANS) attached to either the catalytic domain (Cys-707) or the essential light-chain (Cys-177) of the myosin S1, and acceptor (IAF) in the actin filament (Cys-374) in rigor acto-myosin complexes at 20 oC. When the donor molecule was placed at the Cys-707 of the S1 the distance of the donor from acceptor on the closest monomer was found to be 5.2 ± 0.2 nm with the distributional width of less than 0.5 nm. In the case of the essential light-chain the mean distance between the actin filament and Cys-177 was found to be 7.5 ± 1.0 nm, and the width of the distance distribution was 5.5 ± 1.0 nm. Alternatively, the flexibility of the acto-myosin-subfragment-1 complex was characterized between the catalytic and light-chain-binding domains by measuring the temperature profile of the normalized fluorescence resonance energy transfer, f ' (Somogyi et al., 1984; Biochemistry 23: 3403-3411). In accordance with the simulation data, the protein matrix between the Cys-374 (in actin) and Cys-707 (in S-1) was found to be substantially rigid, while the light-chain-binding domain (Cys-177) experienced substantial flexibility. These results indicate the importance of the dynamic properties of the S1 domains and might provide new insights into the molecular rearrangements in the acto-myosin complex during the muscle contraction.
L-18
NMR STUDIES OF METABOLIC PROCESSES
Zoltán Berentea, Erzsébet Õszb, Balázs Sümegia
aInstitute of Biochemistry, University Medical School
of Pécs
bInstitute of Medical Chemistry, University Medical School
of Pécs, H-7624 Pécs, Szigeti u. 12., Hungary
Nuclear magnetic resonance (NMR) spectroscopy is a powerful instrumental analytical technique, and despite its shortcomings (e. g., low sensitivity, limited number of observable nuclei) it is gaining more and more applications in life sciences. This is probably due to the favorable properties of the method: it is isotope selective, the placement of a signal in the spectrum is very sensitive to chemical environment of the nucleus it is coming from, and in addition, the method is nondestructive and noninvasive. Under certain conditions, the abundance of the signals are proportional to the number of nuclei present in the species they represent, thus, quantitative analysis of mixtures becomes possible. Furthermore, kinetic studies can be carried out involving species with lifetimes over 10-3-10-2 s. Last but not least, radio frequency signals - similarly to X-rays - are not (or weakly) attenuated in living systems (cell suspensions as well as human body), so data can even be acquired in vivo, i. e., the sample can be the living system itself, thus, the information obtained is the most direct possible.
In the lecture we wish to give an overview of the NMR methods commonly
applied in the studies of metabolisms of various living systems, and present
some examples from the projects currently running at our University. These
examples will include the study of the tricarboxylic acid cycle in yeast
and in rodents by isotopomer analysis of 13C 1D NMR or 1H-13C
2D NMR spectra, monitoring of energy metabolism and phosphorylation processes
in yeast in vivo and rat heart extracts by 31P NMR, as
well as enzyme activity measurements.
ABSENCE OF HLA-G PROTEIN ON MACROPHAGES IN FIRST TRIMESTER DECIDUA
S. Trummer1,2*, P. Sedlmayr1, K. Wascher1,2, A. Hammer1, A. Blaschitz1,
W. Walcher3, R. Wintersteiger2 and G. Dohr1
Institutes of Histology and Embryology1, Pharmaceutical Chemistry2
and Department of Obstetrics and Gynecology3, Karl-Franzens-University,
Graz, Austria
HLA-G expression on potential target cells directly and indirectly modulate inhibitory or stimulatory pathways in NK cells. Monocytes and monocytic cell lines have been reported to express HLA-G following stimulation with IFNg which is enhanced by costimulation with GM-CSF or IL-2. Reciprocal stimulation of NK-cells (producing IFNg and GM-CSF) and macrophages (producing IL-12) in the decidua may contribute to early embryo loss.
FACS-analysis of macrophages from healthy first trimester decidua prepared by mechanic dissection, enzymatic digestion and density gradient centrifugation, led to the conclusion that these cells do not express HLA-G protein on the cell surface. In parallel, double fluorescence studies of acetone fixed cryosection of the decidua on a laser scanning microscope had the same result. Further studies are planned to investigate whether decidua macrophages under cytokine stimulation which may appear during pathological conditions are able to express HLA-G.
Key words: macrophages, pregnancy, HLA-G
OPTIMIZATION OF A FAST DERIVATIZATION PROCEDURE FOR VOLATILE CARBONYL COMPOUNDS AFTER TRAPPING ON TENAX
S. Strassnig*, E.P. Lankmayr
Institute for Analytical Chemisty, Micro- and Radiochemistry, Technical University of Graz, Technikerstrasse 4, 8010 Graz
Carbonyl compounds are contained in a wide variety of naturally occuring products and may e.g. contribute essentially to the characteristic aroma of food or fragrant products, even when they are contained in trace concentrations. Depending on their individual composition they can also be made responsible for off-flavours in several matrices as cellulose based packaging materials for example. Thus the determination of volatile carbonyl compounds in liquid or solid matrices is extensively used to assay lipid peroxidation processes or other oxidation reactions.
A method for the determination of carbonyl compounds following a volatilization from liquid or solid samples after trapping on TENAX is presented. Succeeding solvent desorption the carbonyls are derivatized using O-(2,3,4,5,6-pentafluorobenzyl)-hydroxylamine. Reaction occurs in a microwave oven using closed vessels to minimize reaction time compared to state-of-the-art methodology. After gas chromatographic separation of the corresponding oximes they are detected by electron impact mass spectrometry in single ion monitoring mode. Quantification is carried out using internal standardization with 3-fluorbenzaldehyde. The resulting limits of detection are in the ppm-range following the calibration graph method.
The applied conditions yield recoveries which are comparable to existing methodology. Special emphasis was given to an optimization of analysis time. This method has been applied for a determination of volatile carbonyl compounds in heated pure fatty acids, vegetable oils and cardboard so far.
ANALYSIS OF POLYALCANOATE COPOLYMERS BY MEANS OF PYROLYSIS GAS CHROMATOGRAPHY
F. Stelzer*, R. Bona and G. Braunegg
Institute for Chemistry and Technology of Organic Materials, Technial University Graz,
Stremayrgasse 16, A-8010 Graz email:stelzer@ ictos.tu-graz.ac.at and
Institute for Biotechnology, Technical University Graz, Petersgasse 12, A-8010 Graz
If polyalcanoates are produced by biotechnological methods with the help of microorganisms it is still a problem to follow the production of polymer (e.g. polyhydroxybutyrate, PHB). The homopolymer PHB is a crystalline, brittel material but it is well known that copolymerisation with other (e.g.-hydroxyvaleric acid ester, PVA) the thermal behaviour can be changed from hard and brittle thermoplastic to a soft elastomer.
Until now the only method to detect the incorporation of comonomers into the polymer fromed during the fermentation was the gas chromatographic analysis after methanolisation of the polymer. Before this could carried out, the biomaterial (e.g.cell membrane etc.) had to be separated. This was a complicates and time consumming process, results were obtained too late in order to adapt the process to adjust the polymer properties.
In this poster we will show that the copolymer can be well determined by means of pyrolisischromatogrphy. Various copolymers (polyhydroxybutirates-covaleriates)were investigated and two different methods were compared with respect of speed, reproducibility and accuracy.
END GROUP MODIFICATION OF POLY(ETHYLENE GLYCOL)S: ELECTROSPRAY IONISATION MASS SPECTROMETRY AS ANALYTICAL TOOL FOR SUBSEQUENT OPTIMISATION OF REACTION CONDITIONS
G. Schwarzenbacher*, C. Mirtl, M.S. Gangl, M. Goriup and R. Saf*
Institute for Chemical Technology of Organic Materials, Technical University Graz, Stremayrgasse 16, A-8010 Graz, Austria
Tel.: ++43 316 873 8959 Fax: ++43 316 873 8951 e-mail: saf@ictos.tu-graz.ac.at; schwaba@ictos.tu-graz.ac.at
The importance of electrospray ionisation mass spectrometry (ESI-MS) for the characterisation of synthetic monomers, oligomers, and polymers has markably increased in recent years. ESI-MS offers the possibilities to determine molecular weights as well as molecular structures very exactly. Under certain conditions even the mass distribution can be determined. End group analyses, which can be very difficult with conventional methods, are often simplified [1].
The modification of end groups of polymers is an important way to new materials. However, in the case of incomplete conversion, the purification is usually difficult or nearly impossible. Thus, the modification reactions have to be performed as uniform and quantitative as possible - usually optimisation of reaction conditions are required. The most important aspect for an optimisation is the identification of all products formed during the modification, side products included.
We will report on the end group modification of poly(ethylene glycol)-monomethylether, according to Scheme 1. The progress of the reaction and the formation of side products were determined by ESI-MS. An exact and rapid reaction control was possible. Considering the results of the ESI-MS investigations, an optimisation of the reaction conditions as well as of the workup procedures could be carried out.
Refs: [1] R. Saf, C. Mirtl, K. Hummel, Acta Polymer., 1997, 48, 513.
The financial support by the Austrian Science Fund (SFB Elektroaktive Stoffe, Project 10712-CHE, ESF-Program Molecular Magnets) is gratefully acknowledged.
CONVERGENT BEAM ELECTRON DIFFRACTION FOR THE DETERMINATION OF THE TETRAGONAL DISTORTION OF CUBIC CRYSTALS, DEMONSTRATED ON "LOW TEMPERATURE GROWN" GaAs
C. Schür*, M. Leicht, T. Marek and H.P. Strunk
Institute for Microcharacterisation, Friedrich-Alexander-University Erlangen-Nuremberg,
Cauerstr. 6,D-91058 Erlangen, Germany, email:schuer@ww.uni-erlangen.de
Galliumarsenide grown by molecular beam epitaxy (MBE) under arsenic rich conditions at reduced substrate temperature ('low temperature grown' LT-GaAs) is considerably non-stoichiometric. Its excess As content of up to 1.5 atomic % leads to a tetragonal distortion of the epitaxial LT-GaAs layer.
Our motivation is to study the correlation between the microstructure of LT-GaAs and the interesting electro-optical properties of this material, like e.g. the ultrashort lifetime of photogenerated carriers or the high resistivity after annealing.
We use convergent beam electron diffraction (CBED) to determine the tetragonal distortion of these LT-GaAs layers with high spatial resolution. By comparing experimentally acquired electron diffraction patterns (Fig.1) with according numerical simulations (Fig.2), we achieve quantitative results. The chosen orientation of the specimen with respect to the electron beam optimizes the sensitivity of our measurements as well as the application of the theory of dynamical diffraction for the simulations does.

Fig. 1 Experimental diffraction pattern from the LT-GaAs layer, central (000) diffraction disc, (120) zone axis orientation, Ua = 200kV. |

Fig. 2 Dynamical Simulation of the central (000) diffraction disc, (120) zone axis orientation, Ua = 198.9kV, c/c = 0.4. |
THERMODYNAMIC CHARACTERISTICS OF DISPERSION-TYPE PROCESSED CHEESES MADE WITHOUT PEPTIZATION BY DSC METHODS
Balázs Schäffer, Dénes Lõrinczy, Béla Schäffer*
Biophysical Department of University Medical School of Pécs, Szigeti u. 12.,
Pécs, H-7643, HUNGARY
Hungarian Dairy Research Institute*, Tüzér u. 15., Pécs, H-7623, HUNGARY
Besides the traditional method of cheese processing, where Ca breaks down from the protein chain and protein is peptized, a new technology has been elaborated, during which cheese is dispergated without phosphate-containing processing salt, then the gel is formed by plant hydrocolloids.
During our research we used the same composition raw material, one part of which was processed by phosphate-containing processing salt and the other part in the presence of hydrocolloids. Thermodynamic processes taking place, on the one hand, during the processing and, on the other hand, in the finished products were examined by an ultra-sensitive micro DSC-method. The main conclusions are summerized hereinafter briefly.
The samples made with the different processes in the temperature range of 0-40 °C of which the exotherm melting process of incorporated fat taking place show similar shape. On the DSC-curves can be well distingqueshed the two milkfat fraction melting between 0-25 °C and 28-40 °C, from among which with the lower melting point also devides into two fractions well separable. It well indicates that technological step where the fat previously went through on a slow cristallization process so the individual fractions were separately cristallized. At samples, the heat-treatment (processing) of which was carried out in the DSC-equipment, the temperature range of the endothermal process indicating the transformation of protein and hydrocolloids, respectively can be distinguished well. It is 81-90 °C at peptization-type processing and between 61-72 °C at processing without peptization. The differences decrease in the end products, the temperature range is 75-87 °C in traditional processed cheese and 68-74 °C in processed cheeses made without peptization. On the basis of corrigated baselined DSC-curves with the help of PeakFit program it can be proved that the endotherm effect is minimal in the case of heat treatment without peptization, a little bit bigger in the end product than in the heat treated cheese made with peptization and the effect is bigest during the process of peptization. On the basis of results it can be established with a simple DSC-measuring that the heat-treatment was done with or without peptization.
QUANTIFICATION OF THE BLEACHING AGENT "FAS AND ITS DECOMPOSITION PRODUCTS BY HPLC
In pulp and paper industry, FAS (formamidinesulfinic acid) is widely used as a chlorine free bleaching agent. For the development of a biological wastewater treatment process dealing with high loads of FAS and its decomposition products it was necessary to establish analytical techniques for the quantification of these substances.
As organic oxidation and decomposition products of FAS, formamidinesulfonic acid, urea, cyanamide, dicyandiamide, formamidine and an unidentified compound have been reported. These substances show a wide range of different behavior in a biologic treatment process: While urea is known to be highly biodegradable, cyanamide and dicyandiamide are substances of low biodegradability and high nitrification inhibition.
To get a first qualitative information about the composition of the wastewater sample, a thin layer chromatographic (TLC) method was developed on the basis of silica as stationary phase using dichloroethane - methanol (70 : 30) as eluent.
Due to the high polarity of the analytes, the best results for quantitative analysis could be achieved with HPLC using ionexchange resins as stationary phase. It could be shown that both anion and cation-exchange columns can provide effective separation.
For routine analysis a method was optimized on the basis of a sulfonated styrene/divinylbenzene resin loaded with Ca - counterions and an aqueous Ca-sulfate solution as eluent. UV-detection at short wavelength (195 nm) was used. To discriminate between the two coeluting analytes thiourea and FAS, a second wavelength (235 nm) was detected at which the latter has an absorption coefficient close to zero.
HOMOPOLYMERS, STATISTICAL COPOLYMERS AND BLOCK-COPOLYMERS OF NORBORNENE DERIVATIVES: CHARACTERISATION BY MEANS OF CHROMATOGRAPHIC METHODS
Harald Reisinger*, Karin Viertler, Alf Wewerka, Peter Preishuber-Pflügl and Franz Stelzer
Institute for Chemistry and Technology of Organic Materials, Technical University Graz, Stremayrgasse 16, A-8010 Gratz Email: Stelzer@ictos.tu-graz.ac.at
Derivates of norbornene and other cycloolefins with highly diffeerent polarities were polymerized via ring metathesis polymerisation (ROMP). As thisreaction can be concidered to be a living polymerisation, homopolymers statistical copolymers and well definied block copolymerscan be synthetised with narrow molar mass distributions. The chemical composition of the different units and segments varies from pure hydrocarbons to esters, amides, cyano substitued mesogenes and ethers to alcohols and free carboxylic acids.
The characterisation of such copolymers is generally a problematic task, if monomer units of different rigidity and polarity are used it an almost insolvable task.
Our approach to solve these problems is to synthetise well defined standard polymers and to apply various detectors to evau;ation of size exclusion chromatogrammes. Some examples are given with detailed evaluation of data using three different coupled detection system (RI and UV, viscosity and RI or multi angle laser light scattering and RI). The data obtained with the various systems are compared, the different results are discussed, the possibility of getting real absolute molar madd numbers is discussed.
GC-MS INVESTIGATION OF ISOMERIC E- AND Z-4-ARYLIDENE-3-ISOCHROMANONES
Zoltán Pintér1* and Tamás Lóránd2
1Laboratory for Environment Protection, Csárdakörnyék 6, H-7673Pécs, Hungary
2Department of Medical Chemistry, University Medical School of Pécs, Szigeti ut 12.,H-7624
Pécs, Hungary
Recently some new E- and Z-4-arylidene-3-isochomanones have been prepared by us using aldol condensation. These reactions provided either the E- or the Z-isomer or their mixture (see the reaction scheme). A suitable separation method has been found to resolve the diasteroisomers.
In order to investigate purity and thermal stability of the compounds and to identify the structure GC-MS analysis has been performed. In the case of 16 -containing OH-group- we used derivatization reactions to change the chromatographic properties (methylation, silylations with BSA and MTBSTFA). We applied Cool On-Column injection to prevent the molecules thermal degradation during the injection and some cases we applied high temperature split-splitless injections. We did not find decomposition under high temperature injection.
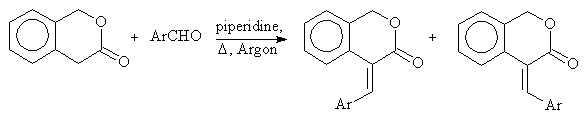
E-series
Z-series
| Compound | Ar | Isomeric comp.(%) | GC-MS studies |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SYNTHESIS AND CHARACTERIZATION OF STEROIDAL CROWN ETHERS
Andrea Petz a*, Zoltán Tuba b , Zoltán Pintér d, Zoltán Berente c,d and László Kollár a,d
a Janus Pannonius University, Department of Inorganic Chemistry, Ifjúság u. 6. (P.O.Box 266) H-7624 Pécs
b Chemical Works of Gedeon Richter Ltd., Budapest
c Institute of Biochemistry, University of Medical School, P. O. Box 99, H-7643 Pécs
d Research Group for Chemical Sensors of the Hungarian Academy of Sciences, P.O. Box 266, H-7624 Pécs
Palladium-catalysed aminocarbonylation of steroidal enol triflates proved to be a very efficient method for the synthesis of steroidal amides possessing high 5a-reductase inhibitor activity 1. On the other hand, the crown ethers of various ring size are well-known complexing agents for alkaline metals. These compounds represent one of the most important groups of chemical sensors 2. Some amino biscrown ethers and crown ethers substituted by an amide functionality (functionalised with amino acid residue) were found to exhibit a highly selective transport of potassium ion and amino acid derivatives through liquid membrane 3,4.
17-Iodo-androst-16-ene (1), 17-iodo-4-aza-androst-16-en-3-one
(2), 17-iodo-6a-hydroxy-3a,5a-cycloandrost-16-ene (3) and
17-iodo-3-methoxy-estra-1,3,5(10),16-tetraene (4) were reacted with
carbon monoxide and 2-(aminomethyl)-15-crown-5 (a) (or 2-(aminomethyl)-18-crown-6
(b)) in DMF in the presence of palladium-phosphine catalysts.
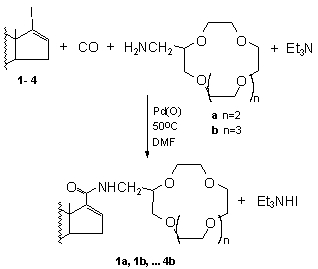
The corresponding steroidal crown ethers (1a, 1b, ....4b) as well as their aza-crown analogs were synthesised in excellent isolated yields and characterized by various NMR techniques and MS.
References
1. Dolle, R.; Schmidt, S.J.; Kruse, L.I. J. Chem. Soc. Chem. Commun. 1987, 904.
2. Bühlmann, Ph.; Pretsch, E.; Bakker, E. Chem. Rev. 1998, 98, 1593.
3. Matsushima, K.; Ikeda, I.; Kohmoto, K.; Nakatsuji, Y.; Okahara, M. J. Jpn. Oil Chem. Soc.
(Yukagaku) 43, 556 (1994); Chem. Abstr. 121, 205327a (1994).
4. Inokuma, S.; Matsunaga, S., Negishi, T., Hayashe, T., Kuwamura, T., J. Jpn. Oil Chem. Soc.
(Yukagaku) 38, 170 (1989); Chem. Abstr. 111, 25347s (1989).
SPME - A SIMPLE SAMPLE PREPARATION METHOD FOR MIGRATION STUDIES
K. Lübke*, E. Leitner, W. Pfannhauser
The SPME device consists of a polymer-coated fused-silica fibre connected to a stainless steel tubing. Both are contained in a syringe needle for protection, with the steel tubing serving as a plunger to move the fibre up and down. When the fibre is withdrawn into the needle, the latter can be used to pierce a septum, then the plunger is pressed down to expose the fibre to the sample or its vapours. After exposition the fibre is withdrawn again and the SPME device can be put into the injector of a GC with a specially designed inlet liner, where the analytes are desorbed rapidly when the fibre has been pushed down.
In the present work different types of fibre coatings were compared and optimised for our application. We have been working with 7µm poly(dimethylsiloxane) (PDMS), 30µm PDMS, 65µm PDMS/divinylbenzene (DVB), 75µm Carboxen (small carbon particles embedded in PDMS) and 50/30µm DVB/Carboxen/PDMS. Analysis was performed by gas chromatography/mass spectrometry (GC/MS). The chromatograms obtained show a broad, hill-like signal over several minutes, corresponding to a large number of different volatile substances. Despite the use of a MS detector, only a few compounds could be resolved and identified. Therefore we used deuterated dodecane as internal standard for calculation of the total amount of volatile compounds as dodecane equivalents. The ratio of the integrated signal areas of internal standard and analytes, respectively, shows good reproducibility, but varies with the coating material. The sensitivity for the analytes also strongly varies with the fibre coating used.
REFERENCES:
[1] J. PAWLISZYN, Solid Phase Microextraction. Theory and Practice, 1997, Wiley
[2] Zh. ZHANG, M. J. YANG, J. PAWLISZYN, Anal-Chem. (1994) 66(17), 844A-853A.
[3] T. NILSSON et al, J. High Resolution Chromatogr. (1995) 18, 617-624.
DETERMINATION OF ESTRIOL IN HUMAN PLASMA BY STABLE ISOTOPE DILUTION GAS CHROMATOGRAPHY-NEGATIVE ION CHEMICAL IONIZATION MASS SPECTROMETRY.
H.J. Leis*, G. Raspotnig, W. Windischhofer, G. Fauler
University Children's Hospital, Department of Analytical Biochemistry and Mass Spectrometry, Auenbruggerplatz 30, A-8036 Graz, Austria
A stable isotope dilution gas chromatography-mass spectrometry (GC-MS) assay for the trace level determination of estriol in human plasma is desrcibed. Negative ion chemical ionization (NICI) mass spectrometry is used for highly specific detection. The method involves derivatization of the phenolic hydroxyl to the pentafluorobenzyl ether derivative and subsequent reaction of the remaining hydroxyls with heptafluorobutyric anhydride. This derivative allows detection of the strikingly abundant phenolate ion under NICI conditions. 2H3-labeled estriol was used as an internal standard. For high level measurements (>312 pg/ml) plasma was directly derivatized without any preparation step. A rapid and simple sample work up procedure was elaborated for trace level determinations (>5 pg/ml plasma) using solid phase extraction on C18 with a recovery of 92.9%. For low level measurements, the calibration curve was linear in the range of 4,88 to 625 pg/ml (r=0,99993). Inter-assay analytical precision (CV) was 1,29%, 2,30% and 2,89% at 39,06, 156,25 and 650 pg/ml plasma, respectively. For high level measurements, calibration curve linearity was observed in the range of 0,312 to 20 ng/ml (r=0,99998). Inter-assay analytical precision (CV) was 5,17%, 1,92%, 2,57% and 2,74% at 0,312, 0,625, 2,5 and 10 ng/ml plasma, respectively. Sensitivity and specificity of the presented method allows adequate determination of estriol in human plasma samples.
ENANTIOSEPARATION OF BETA-BLOCKERS, SYMPATHOMIMETICS AND HYDROXY ACIDS BY LIGAND EXCHANGE CAPILLARY ELECTROPHORESIS
O. Lecnik*, M. G. Schmid and G. Gübitz,
Institute of Pharmaceutical Chemistry, Karl-Franzens-University Graz,Universitätsplatz 1, A-8010 Graz, Austria
Ligand exchange capillary electrophoresis has successfully been applied to the direct resolution of underivatized amino acids using L-proline, L-4-hydroxyproline and N-alkyl-L-4-hydroxyproline in form of their copper(II) complexes as chiral selectors [1, 2]. This contribution deals with the application of this basic approach for the enantiomeric separation of drugs containing amino alcohol structure and hydroxy acids.
Copper(II) complexes of N-alkyl-L-4-hydroxyproline such as N-propyl- and N-octyl were compared for their ability of resolving amino alcohols and hydroxy acids.
Several drugs containing amino alcohol i. g. sympathomimetics and ß blockers were resolved into their enantiomers. Hydrophobic interactions of the lipophilic chain of the selector and the alkyl groups of the analytes might enhance selectivity. The influence of the concentration of the chiral selector, the copper(II) content, the electrolyte composition and the pH as well as the addition of organic modifiers on the enantioselectivity was investigated. For the separation of hydroxy acids the pH optimum was found to be 4.3, whereas amino alcohols were optimally resolved at pH 12. Since in HPLC it is not possible to work at high pH, CE is advantageous for the chiral separation of this compound class using the principle of ligand exchange.
[1] M.G. Schmid and G. Gübitz, Enantiomer, 1, 23-27 (1995)
[2] Á. Végvári, M.G. Schmid, F. Kil·r and G. Gübitz, Electrophoresis,19, 2109-2112 (1998)
SURFACE CHARACTERIZATION OF PHOTOCHEMICALLY MODIFIED POLYMERS
Wolfgang Kern1*, Ursula Meyer1, Thomas Kavc1, Eva-Maria Hoiser1, Maria F. Ebel2, Robert Svagera2 , Peter Pölt3
(1) Institut für Chemische Technologie organischer Stoffe, Technical University of Graz,
Stremayrgasse 16, 8010 Graz, Austria
(2) Institut für Angewandte und Technische Physik, Vienna University of Technology,
Wiedner Hauptstraße 8-10, 1040 Wien, Austria
(3) Forschungsinstitut für Elektronenmikroskopie und Feinstrukturforschung, Technical University of Graz, Steyrergasse 17, 8010 Graz, Austria
During the last years, the chemical and physical properties of polymer surfaces, such as chemical composition, hydrophilicity, conductivity and crosslink density, have attracted attention. Since specific properties of the surface are required for numerous applications, several techniques for the modification of polymer surfaces have been developed. Wetchemical etching, plasma treatments and corona discharges are well established. In addition, the grafting of vinyl monomers onto polymer surfaces utilizing radiation (UV, e-, ) has been successfully applied.
Our aim was to attach specific functional groups onto polymer surfaces employing reactive gases in the presence of UV radiation. Compared to plasmachemical techniques employing reactand gases, a photochemical way appeared promising as it operates under atmospheric pressure and does not require expensive setups.
Part of the present work is concerned with the introduction of sulfonic acid groups onto polyethylene (PE) surfaces. PE samples (LDPE and HDPE) were irradiated with UV light (Hg lamp) in a gas atmosphere containing SO2 and O2 to achieve a photosulfonation of the surface. On the other hand, basic and neutral groups were attached onto polystyrene (PS) surfaces employing photoreactions. UV irradiation of PS in the presence of hydrazine vapours led to the modification with NH2 groups, with gaseous BrCN surfaces modified with CN groups were obtained.
Further work was done in order to derivatize the functional groups introduced. E.g., the conversion of CN groups to amides and carboxylic acids is described. In a similar way, sulfonic acid groups were converted into sulfonamides.
All modified polymer surfaces were characterized by means of FTIR and ATR-FTIR, REM-EDX and X-ray photoelectron spectroscopy (XPS). In addition, the presence of ionizable groups was evidenced by dyeing with a cationic dye (methylene blue). In order to investigate the wetting behaviour of the surfaces, contact angles of water were measured
Summing up, the presentation is concerned with the modification of polymer surfaces employing UV radiation and with surface analytical investigations.
COMBINED TLC AND CAPILLARY GAS CHROMATOGRAPHIC METHOD FOR THE QUANTIFICATION OF PHYTANIC ACID AND VERY LONG CHAIN FATTY ACIDS IN SERUM OF PATIENTS WITH PEROXISOMAL DEFICIENCIES
Susan Juricskay
Central Research Laboratory, The University Medical School of Pécs, 1 Honvéd u. Pécs, Hungary, H-7643
Peroxisomes were initially designated by Rodin in 1954 who noted spherical oval bodies with a single limiting membrane and a fine granular matrix in mouse kidney. The importance of peroxisomes in cellular processes became evident with the discovery of enzymes for the b-oxidation of fatty acids, especially very-long -chain fatty acids(VLCFAs), i.e., carbon chain length greater than 22.
The peroxisomal diseases are inherited metabolic diseases, which can be characterised by a build-up of metabolites normally degraded by peroxisomal enzymes or by decreased amounts of metabolites normally synthethised by peroxisomes, depending on the specific enzymatic defects, produce severe neurodegenerative effect.
Peroxisomal diseases can be classified into three groups based on single or multiple defects and integrity of peroxisomes. As phytanic acid and VLCFAs are very important in the differential diagnosis of the peroxisomal diseases, it is an essential and reliable method for their determination.
The following method was adopted for TLC:
Fatty acid analysis: Extraction and methilation:Internal standards of 100 nmol pentacosanoic acid(C:25)and 2 nmol of hexacosanoic (C27) were added to 1 ml serum. After extraction with nine volume chloroform : methanol (1:1) and centrifugation, the died sediment was dissolved in 1N methanolic HCl and was incubated overnight at 75 oC.
TLC: Chromatograms were developed on 20 x 20 cm silica-gel plates with hexane : diethylether : acetic acid (90:30:1).
GC analysis: was performed on a Hewlett-Packard 5890 GC in splitless mode on a ULTRA-1 ( Crosslinked Methyl Silicone Gum) column (25 m x 0.33 um x 0.2 mm and a HP-INNOWax (Crosslinked Polyethylene Glycol) column( 30m x 0.5 um x 0.32 mm ) column. Samples were dissolved in hexane.
Temperature program was as follows: initial temperature of 70oC was hold for 2 min, then increased to 160 oC at 10 oC/min. After a 4-min isotherm period the temperature was raised to 270 oC at 5 oC/min. After a 1 min isotherm period it was increased to 300 oC by 20 oC/min and held for 2 min.
Results: The normal values were calculated from 10 samples of healthy individuals. Serum of 40 patients were investigated, in the case of 16 patients phytanic acid increased and in 4 patients C:26 increased. One patient had Zellweger syndrome and she died before her first birthday.
Conclusion: The applied method is suitable for determination of phytanic acid and VLCAs and it gives an useful help in the differential diagnosis of peroxisomal diseases.
DEVELOPMENT OF RP-HPLC METHOD FOR DETERMINATION OF SELECTED GROUP OF PESTICIDES IN SOILS
Milan HUTTA, Mária CHALÁNYOVÁ, Róbert GÓRA*, Radoslav HALKO, Marek PAJCHL a S. DOKUPILOVÁ
Department of Analytical Chemistry, Faculty of Natural Science, Komensky University,
Mlynská dolina CH-2, SK-842 15 Bratislava, Slovak Republic E-mail: gora@ fns.uniba.sk
Development of validated method for routine determination of pesticide residua in soil is in the case of wide and chemically miscelaneous group of pesticides influenced by:
CHARACTERIZATION OF SOIL HUMIC SUBSTANCES BY LIQUID CHROMATOGRAPHY METHODS
Róbert GÓRA*, Milan HUTTA, Ján KANDRÁÈ
Department of Analytical Chemistry, Faculty of Natural Science, Komensky University,
Mlynská dolina CH-2, SK-842 15 Bratislava, Slovak Republic E-mail: gora@ fns.uniba.sk
Humic substances (HS) create a complex mixture of macromolecular refractory organic substances combined to structural entities which probably even have no two molecules identical. We can find them at various places in our environment ( soils, sediments, waters, dust particles, peats, coals etc.)
Almost all their properties are dispersed and are tightly bound to their structural polydispersity, polarity, size etc. However, their properties depend also on procedures used for their isolation from particular matter, place of origin, climate acting during their constitution and residence.
Their composition and constitution influences their complex-forming ability, solubility in various solvents, aggregation susceptibility and redox properties beside many other features. Contrary to other chemical individuals we should accept opinion that humic substance ( humic acid, fulvic acid etc.) is always "variation to the theme" and not the individuum.
Methods of liquid chromatography play important role mainly in the fields of
In presented work some of mentioned aspects and associated problems of LC application of HS containing samples are discussed.
METHOD DEVELOPMENT FOR BROAD RANGE SCREENING AND SIMULTANEOUS DETERMINATION OF BIOCIDES IN RIVER SAMPLES FROM EASTERN CHINA
Marion Gfrerer*, Thomas Wenzl, Ernst Lankmayr
Institute of Analytical Chemistry, Radio- and Mikrochemistry, Technical Univeristy of Graz, Technikerstr. 4, A-8010 Graz, Austria
Owing to the persistence and toxicology of polychlorinated organic compounds (PCOC's) and other biocides their environmental impact to surface water by agricultural and industrial activities represents a considerable risk to animals and human beings, especially when contamination of our most important resource - drinking water - occurs.
Within the frame of a co-operative research project, which is supported by the European Union, a risk assessment for the waters of the Liao- and Yangtse-River (Eastern China) should be established [1]. Therefore, the principle goal was the development of analytical methods for a simultaneous extraction and determination of PCOC's from river samples. Water samples, suspended particulates and sediment samples of the two rivers were investigated with respect to their content of 21 common polychlorinated biocides and metabolites.
For the purpose of method development test-samples spiked with PCOC's were prepared. These were used to establish recoveries and relative standard deviations especially for the extraction and clean up procedures. The analytes were extracted from the matrices by microwave-assisted-extraction (MAE), reversed phase solid-phase-extraction (SPE) and fluidized-bed-extraction (FBE). Clean up and concentration was performed by silica-gel-chromatography. Finally, the determination of the specimen was performed by gas chromatography combined with mass selective detection (GC-MS).
GC-MS analysis was carried out under various aspects. Our first goal was the quantification of selected target compounds - all of them PCOC's - , which was performed in selected ion monitoring (SIM) mode. For these compounds the limits of detection were calculated statistically. The second aim of the studies was a screening of the samples for other biocides, which were not part of the priority list, which was performed in SCAN mode. Spectral data of the samples were compared with library data and retention time indices were calculated to ensure the results.
As a result, the concentrations of the 21 organochlorine target analytes, i.e. PCBs, HCHs, DDT and Cyclodienes are rather low and are not threatening the water quality of the river systems. Nevertheless, even at low concentration a considerable mass transport may take place in case of the Yangtse-River. Some evidence was found that other compounds might be a problem in the Liao-He. For example, atrazine was found in the scan-mode. Finally, the results for the 21 target compounds of the EU-Project were compared with those of the project partners [2].
In on-going studies, new substance classes, such as organophosphorous pesticides and triazines, are added to the PCOC's and are SIM-quantified further on. As a first result, atrazine and its metabolite desethyl-atrazine was found in relatively high concentrations in the water of the Liao river.
[1] B. Gawlik and B. Platzer, Investigation and Risk Assessment of Polychlorinated Organic Compounds (PCOCs) in Drinking Water Resources of Developing regions in Eastern China - Report on the Kick-off-Meeting, INCO-DC ERBIC18CT970166, 1998
[2] B. Gawlik, B. Platzer and H. Muntau (Editor), On the Presence of Polychlorinated Organic Compounds in the Liao River and Yangtse River in Eastern China - Pilot-sampling campaign - Final report, EUR 18702 EN, 1999
METAL ASSOCIATIONS IN CARIOUS TEETH OF INDUSTRIAL AREAS OF UKRAINE
1Boyko T.* and 2Kosmus W.
1- Institute of Geology and Geochemistry of Combustible Minerals, National Academy of Sciences of Ukraine, Naukova Sr., 3a, 290053, Lviv, Ukraine, boyko_t@yahoo.com;
2- Institute of Analytical Chemistry, Graz University, Universitaetsplatz, 1, A-8010, Graz, Austria, walter.kosmus@kfunigraz.ac.at
Performing this study was caused by recently arisen local stomatological problems in the industrial areas of Lviv region associated with Lviv-Volyn coal mining activities as well as exploitation of the Precarpathian native sulphur deposits. Collected children's permanent teeth were digested using Microwave Digestion System (MLS 1200 MEGA). Analyses for metal concentration were carried out using Inductively Coupled Plasma Mass Spectrometer (ICP-MS). Caries disease is followed with the increase of the major metals concentration. In all analysed samples, concentration of the main cation of hydroxyapatite - calcium was found to be at the similar level that published earlier for calcified tissue. However it is three times higher then in teeth reported for industrial Polish regions. The concentrations of lithium and strontium have the lowest spread.
Performed principal components analysis separates two antagonistic metal associations: carious resistant and carious supporting. To the first one belong Ca, Sr and Li. Its existence indicates the positive role of Sr and Li on the stability of the tooth enamel. Strontium has already been regarded to be anti-caries agent capable of preventing the dissolution of tooth enamel. However, the biogeochemical role of Li in tooth structure is not completely understood by us at the moment.
Second group of elements is connected with development of caries disease and is divided into two subgroups: Zn, Cd, Cu, Pb, Sn and Ag (a) as well as Ba, Cr, Al, Bi, Mo, Mn, Fe, Co, Be and V (b). Structural disorder in tooth mineral can be induced through the isomorphic substitutions for the main cation and anions. Because the carious teeth were collected from in the region with the sulphur excess in the environment, it was suggested the possibility of the next partial replacement 2P5+=S6+. It could be balanced by cation substitutions of Ca2+ with metals of the subgroup (a) mainly forming the bivalent cations (except Ag): Zn, Cd, Cu, Pb, Sn and Ag. The sorption on the hydroxyapatite surface, as ºPOZn+ or ºPOCd+ could also take place.
Subgroup (b) includes the next clusters: Mn-(Co-Ba), Fe-(Cr-Bi) and Al-(Mo-Be-V). Grouping the anions and cations in one cluster, suggests the coupled substitution. Earlier studies showed that Mn can enter the lattice as (MnO4)- replacing the (PO4)3- groups as well as (Co)2+ and (Ba)2+ - (Ca)2+.
INFLUENCE OF Ti(IV)ASCORBATE ON CARBOHYDRATE METABOLISM IN CADMIUM TREATED WHEAT
Ildikó Kerepesi1, Éva Stefanovits-Bányai2 Zoltán Szabó1, Éva Sárdi2, István Pais2
1Department of Analytical and Structural Chemistry, Janus Pannonius University, H-7601 Pécs, Hungary
2University of Horticulture and Food Industry, Budapest, Hungary
Cadmium in known to be one of the most toxic heavy metals released in the environment. Several studies, mostly in the laboratory, indicate an altered carbohydrate and nitrogen metabolism in plants affected by heavy metal pollution (Kabata-Pendias and Pendias 1985.). During the last few years the intensive investigation of Ti on nutrient uptake and plant metabolism was carried out (Pais 1983).
Water-soluble carbohydrates contributing in the response to Cd++ stress in Ti(IV) ascorbate containing nutrient solution in wheat seedlings were studied. Total water-soluble carbohydrate, glucose, fructose, sucrose glucan and fructan contents, as well as the cadmium and titan content were measured in wheat seedlings exposed to Ti(IV)ascorbate in 10-4 M Cd++ , 10-5 M Cd++ containing medium. Titanium and cadmium content were measured by ICP and carbohydrates by spectrophotometric method.
Our results suggest that there were mainly glucose, fructose and fructan which showed considerable response to Cd, ascorbic acid and titanium treatment. Sugar content in plants exposure to Cd++, increased and this increment was higher in the case of higher metal concentration. Tendentiously, titanium decreased the cadmium-induced sugar accumulation.
Ti(IV)ascorbate and its component ascorbic acid were also applied without Cd++, to study the operation of these chemicals. In general, ascorbic acid induced higher accumulation in measured sugar components than Ti(IV)ascorbate itself did.
Titanium in Cd++ containing solution caused significantly lower cadmium accumulation in leaves, than that of without Ti(IV). Differences in titanium content was observed only in roots, showed significantly higher values in plants grown in Cd++ 10 -4 M contained solution.
These results and discussions are detailed in the paper.
References:
Kabata-Pendias, A. and H. Pendias. 1985. Trace elements in soil and Plants. CRC Press. Boca Raton, Florida.
Pais, I. 1983 Titanium and plant response. Nutri. 6: 3-131.
STUDY ON A CATALYTIC CHEMILUMINESCENCE REACTION
K. Szabóa*, K. Szaláncza, N. Mareka
a Department of Inorganic Chemistry and Technology , Janus Pannonius University
H-7624 Pécs, Ifjúság u. 6., Hungary (e-mail: szabo@ttk.jpte.hu)
It is known, that under reaction of oxalateacid-ester type compounds containing
O O
A-O-C-(C)n-O-B
group (where A and B are aliphatic or aromatic ester groups, and n > 1) with H2O2 an excited compound is produced, which produces chemiluminescence when reacting with fluorophores containing three condensed aromatic groups (e. g. anthracene, naphtacene and some derivatives of them).
The mechanism of the reaction usually involves three main steps:
1, ester + H2O2 intermediate product
2, (intermediate product)* + F F* + intermediate product
3, F* F + h.
The structure of the intermediate product and the mechanism of energy transfer is still not really clear. Our aim was the study this type of reactions. As a model system bis-(2,4,5-trichloro-6-carbo-butoxy-phenyl)-oxalate and H2O2 was choosen. The reaction was catalyzed by sodium-salycilate. The fluorophore was 9,10-dibromo-anthracene. The intensity and the duration of photoemission was maximized as functions of the temperature and the concentration of H2O2 and the catalyzer.
A FLUORESENCE SPHINGOMYELINASE ASSAY USING A CCD CAMERA
A. Loidl*, R. Claus, P. Deigner, A. Hermetter
Department of Biochemistry and Food Chemistry, Technische Universität Graz,
A-8010 Graz, Austria and Institute of Pharmaceutical Chemistry, University of Heidelberg, Heidelberg, Germany
Sphingomyelinases are important enzymes of signal transduction. They catalyze the hydrolysis of the sphingolipid sphingomyelin, giving rise to the intracellular formation of biologically active ceramide and phosphatidylcholine.
Here we report on a fluorescence method for the fast and accurate determination of this enzyme in biological samples. The assay is based on a fluorescent sphingomyelin analogue carrying fluorescent NBD-dodecanoic acid instead of an aliphatic acyl chain at the nitrogen atom. The fluorescent substrate is hydrolysed by cellular sphingomyelinases and fluorescent ceramide is formed which can be separated from the remaining substrate using thin-layer chromatography on silica gel. The fluorescence intensity pattern on the TLC plate thus obtained can accurately be determined using a C.C.D. camera. Typically, a large number of samples can be analysed at a time within seconds. Examples for the quantitative analysis of sphingomylinases from freshly prepared cellular homogenates as well as from commercial sources will be given.
APPLICATION OF SEMICONDUCTOR LIGHT SOURCES FOR TIME-RESOLVED FLUORESCENCE MEASUREMENTS
Stephan Landgraf
Institut für Physikal. u. Theoret. Chemie, Technikerstr. 4/I, A-8010 Graz
Applying semiconductor light sources for the determination of nanosecond fluorescence lifetimes we developed a new experimental technique in our institute [1-2]. Using the modulation method and a digital storage oscilloscope for recording the data it is now possible to investigate chemical systems with an absorption from 370 to 670 nm and a lifetime from 100 ps to 1 µs [3-4]. Since several years it is discussed to use time-resolved fluorescence methods for chemical sensing in environmental or medical applications [5].
After a lot of investigations of several different fluorophores we also decided to determine environmental relevant substances. In crude oil, which can be found in many regions, polycyclic aromatic compounds are present. These components are fluorescent and can be excited with light up to more than 400 nm. The emission is relatively strong and typically shifted by 40 nm to longer wavelength. Measurements at different frequencies show are typical curve, which can be used to characterize the sample as crude oil and to distinguish the origin of the sample. Using ultrabright light emitting diodes (LED) it was already possible to determine the fluorescence lifetime in a solution in cyclohexane down to a concentration in the region of ppm.
The experiments were carried out under inert gas condition as well as under ambient conditions. Air quenches the fluorescence to 70 % of the inert gas values. The shape of the curve (lifetime vs. frequency) is changed due to the different lifetimes in the mixture of fluorescent compounds in crude oil.
It is planned to modify the experimental setup to measure directly on solid samples similar to those, that can be found in environmental applications.
1) S. Landgraf, G. Grampp, J. Inf. Rec. Mats. 23, 203207 (1996).
2) S. Landgraf, G. Grampp, J. Inf. Rec. Mats., 24, 141-148 (1998).
3) J. Sobek, S. Landgraf, G. Grampp, J. Inf. Rec. Mats., 24, 149-156 (1998).
4) T. Fayed, G. Grampp, S. Landgraf, Int. J. Photoenergy, 1999, 1, art. 37.
5) S.B. Bambot, J.R. Lakowicz, G. Rao, Trends Biotechn. 13, 106115 (1995).
FLUORESCENCE MONITORING OF FREE RADICAL-MEDIATED LIPID OXIDATION AND ITS INHIBITION BY ANTIOXIDANTS IN SERUM
H. Brandstätter*, B. Mayer, G. Hofer, A. Hermetter
Department of Biochemistry and Food Chemistry, Technische Universität Graz,
A-8010 Graz, Austria
Free radical-mediated oxidation of biomolecules is a harmful process, which is involved in the development of many diseases. Antioxidants counteract this processes, depending on their lipid or water solubility.
We developed a high-throughput screening method for the analysis of lipid oxidation in blood serum and its inhibition by natural antioxidants. For this purpose, a fluorescent analogue of the main serum phospholipid phosphatidylcholine was used. The respective compound contains a fluorescent fatty acid which shows the same susceptibility towards oxidation as compared to natural (polyunsaturated) fatty acyl chains. Under oxidative stress, the fluorophore is destroyed and from the time-dependent decrease in fluorescence intensity the kinetics of lipid oxidation can be determined. Before the bulk of lipids is oxidized (propagation phase), a lag phase is observed during which the protective antioxidants are consumed. Thus, the length of the lag phase is representative for the antioxidant capacity of serum. We screened a large number of biological samples and antioxidants using a fluorescence plate reader. We found that water- (e.g. Vitamin C) and lipid- (e.g. Vitamin E) soluble vitamins led to a concentration-dependent prolongation of lag times corresponding to an improvement of the antioxidant capacity of serum.
ISOLATION AND CHARACTERISATION OF CAROTENOIDS FROM THE FRUITS OF
ASPARAGUS FALCATUS
Deli J.*, Molnár P., Ôsz, E., Tóth G, 1Haberli, A., 1Pfander, H.
Department of Medical Chemistry, University Medical School of Pécs, Szigeti út 12, H-7643 Pécs Hungary
1Department of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012, Bern, Switzerland
During our investigations of different species of paprika (Capsicum annuum) some novel carotenoids containing 7-oxabicyclo[2.2.1]heptyl (3,6-epoxi-b) end group and 3,5,6-trihydroxy-b-end group have been isolated and characterized [1,2]. These compounds may be formed from antheraxanthin and violaxanthin, and their occurrence may be interrelated with the biosynthesis of k-end group, which has not been clarified in every detail yet.
As a continuation of our work on biosynthesis of carotenoids, we aimed at investigating the other natural sources containing carotenoids with k-end group, and identifying and isolating all existing carotenoids containing a 3,5,6-trihydroxy-5,6-dihydro-b- or 3,6-epoxy-b-end group.
In this present paper, the authors report on the HPLC investigation of the fruits of Asparagus falcatus, and the isolation of the main and minor compounds. Beside the main components (capsanthin, zeaxanthin, b-cryptoxanthin), some minor carotenoids (capsorubin, capsanthin 5,6-epoxide, violaxanthin, mutatoxanthin, capsanthone, 5,6-diepikarpoxanthin) and several Z-carotenoids (13Z- and 9Z-capsorubin, 13Z-, 13'Z-, 9Z-, 9'Z-capsanthin, 9Z-capsanthin 5,6-epoxide, 9Z-violaxanthin) were isolated in crystalline state and characterized by their UV/VIS, NMR, CD and mass spectra.
The stereospecific occurrence, isolation and characterization of 9Z-capsanthin 5,6-epoxide has a considerable importance.
[1] Deli J., Molnár P., Matus Z., Tóth G. Steck A.: Helv. Chim. Acta 79, 1435 (1996)
[2] Deli J., Molnár P., Matus Z., Tóth G.,
Steck A., Pfander H.:Helv. Chim. Acta 81, 1233 (1998)
b) Institute of Biochemistry, University of Medical School, H-7643 Pécs, P. O. Box 99
c) Research Group for Chemical Sensors of the Hungarian Academy of Sciences, H-7624 Pécs, P.O. Box 266
Although the PtX2(L) (X=Cl, Br, I; L= bidentate ligand) complexes are among the most investigated ones and their catalytic importance is steadily increasing, surprisingly little is known about their Pt(CN)2(L)-type analogues. While the various reaction pathways with dppm (bis(diphenylphosphino)methane) furnished [Pt(dppm)(CN)2]2 and [Pt(dppm)(CN)]2 dinuclear complexes containing dppm bridges, which were characterised by x-ray crystallography 1,2, Pt(CN)2(dppp) mononuclear complex was selectively obtained with dppp (1,3-bis(diphenylphosphino)-propane) 3.
Pt(CN)2(Ph2P(CH2)nPPh2) (n=2,3,4) and Pt(CN)2(P1-P2) (Figure 1; P1-P2 = 1-diphenyl-phosphino-2,1'-[(1-diphenylphosphino)-1,3-propanediyl]-ferrocene, 1-diphenyl-phosphino-2,1'-[(1-dicyclohexylphosphino)-1,3-propanediyl]-ferrocene) were synthesised by reacting potassium cyanide and the corresponding PtCl2(diphosphine) complexes. PtCl(CN)(diphosphine) complexes were identified as minor products when KCN to PtCl2(diphosphine) molar ratio was kept below 2. The use of KCN in excess resulted in the formation of K2Pt(CN)4.
[Pt(CN)({Ph2P(CH2)2}2PPh)]+ complex cation and Pt(CN)2)({Ph2P(CH2)2}2PPh) five-coordinate covalent complex of fluxional behaviour (Figure 2) were obtained at KCN/Pt ratio of 1 and 2, respectively. The platinum-cyano complexes were characterised by 31P and 13C NMR. The direct Pt-CN bond was proved by 1J(195Pt,31P), 2J(31P,13C) coupling constants by using sodium cyanide-13C for ligand exchange reactions.
All the Pt(13CN)2(P-P)-type complexes show typical AA'XX' second order 31P NMR spectra. The calculated values of 2J(PA, PA'), 2J(CX, CX'), 2J(PA, PX), and 2J(PA, PX') as well as detailed NMR characterisation of the above complexes will be presented.
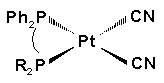
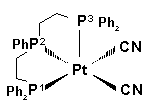
Figure 1 (R=Ph, Cy) Figure 2
References
1. M. N. I. Khan, C. King, J-C. Wang, S. Wang, J. P. Fackler, Jr., Inorg. Chem. 1989, 28, 4656.
2. C-M. Che, V. W-W. Yam, W-T. Wong, T-F. Lai, Inorg. Chem. 1989, 28, 2908.
3. A. R. Khan, S. M. Socol, D. W. Meek, R. Yasmeen, Inorg. Chim. Acta 1995, 234, 109.
INVESTIGATION OF TRANSMISSION OF SUBSTITUENT EFFECTS IN E-2-BENZYLIDENE DERIVATIVES OF CYCLOHEXANONE AND TETRALONE BY IR SPECTROSCOPY AND THEORETICAL METHODS
bDepartment of Organic Chemistry, Komensky University, Bratislava, Slovakia E-mail: perjesi@apacs.pote.hu
Numerous synthetic and natural alpha,beta-unsaturated carbonyl compounds exhibit remarkable biological activity. Their biological effects are considered to depend on their reactivity towards the thiol moiety of the critical peptides and/or proteins. If this assumption is true, then electronic and steric factors should affect the reactivity, i.e. their biological activity.
Recently we have prepared and investigated in vitro antifungal activity of the title E-2-benzylidenecyclohexanones (1) and -tetralones (2) [1]. In order to investigate the electronic and steric structures of the compounds we have have investigated the transmission of substituent efects in the two series by IR spectroscopy.
1 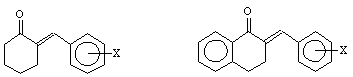
2 X = N(CH3)2, OCH3, H, F, Cl, Br, NO2
The C=O stretching wavenumbers of the compounds were correlated with the Hammett sigma and the Swain-Lupton F and R substituent constants and the results were compared with those obtained for similar correlations with series of related cyclic and acyclic unsaturated componds. The geometry and the electronic structure of the parent compounds of the above series were investigated using MNDO, AM1 and PM3 methods. New qualitative and quantitative relations were found between the IR spectral data, transmission ability, geometric properties and electronic parameters of the investigated systems.
[1] P. Perjési, T. M. Al-Nakib, M. Abdulhamid: Structure-activity relationship and acute toxicity of new alpha,beta-unsaturated ketones. Health Sciences Poster Day, Faculty of Medicine, Kuwait University (Kuwait), April 20, 1998.
FT-IR AND RAMAN STUDY OF SOME NEW E- AND Z-4-ARYLIDENE-3-ISOCHROMANONES
Tamás Lóránd1*, Sándor Holly2 and Gábor Keresztury2
1Department of Medical Chemistry, University Medical School of Pécs, Szigeti ut 12.,H-7624 Pécs, Hungary
2Chemical Research Center Hungarian Academy of Sciences, Institute of Chemistry, Pusztaszeri ut 59., H-1025 Budapest, Hungary
Some new E- and Z-4-arylidene-3-isochromanones have been synthesized starting from 3-isochromanone and aromatic aldehydes by using base catalyzed aldol condensation. The isomeric composition of the products was influenced by the aromatic aldehydes furnishing either the E- or the Z-isomer or their mixture (see the reaction scheme below).
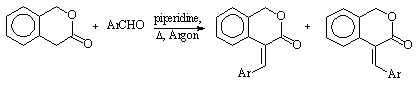
E-series Z-series
|
|
Ar |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The furyl derivative, 13, was chosen for detailed theoretical investigation. Considering four different starting configurations, the structural parameters of the lowest energy conformers of both the E- and Z- isomers were determined by quantum chemical geometry optimization, then the vibrational spectra were predicted by means of the scaled quantum mechanical force field method. The calculations were done according to the Becke3LYP/6-31G* density functional (DFT) method. Comparison of the calculated and measured IR spectra allows a more definitive assignment of the observed bands and provides a deeper understanding of the differences in the vibrational spectra brought about by isomerization and conformational changes.
FT-IR STUDY OF SOME CAROTENOIDS
Tamás Lóránd*, József Deli, Péter Molnár and Gyula Tóth
Department of Medical Chemistry, University Medical School of Pécs, Szigeti ut 12.,H-7624 Pécs, Hungary
Several natural and synthetic carotenoids have been investigated by FT-IR spectroscopy. The IR bands of the characteristic functional groups (CH3, CH2, C=C, C=O, OH, etc.) were assigned if it was possible. Some special functional groups -without hydrogen atoms- as C=C=C, cross epoxides, etc., which can not be easily identified by 1H NMR methods were detected by FT-IR spectra, too.
FLOW INJECTION DETECTION OF UREA USING A PLANAR WAVEGUIDE BIOSENSOR AS DETECTOR
Barna Kovács*, Roland Dombi and Géza Nagy
Research Group for Chemical Sensors, MTA-JPTE, Department of General and Physical Chemistry, Janus Pannonius University, H-7624 Pécs, Ifjúság u. 6, Hungary
Optical sensors based on absorption measurement are widely used for the determination of biologically active molecules. The use of planar waveguide sensors has two distinct advantages: i) the sensing layer could be thin which results in a short response time, and ii) the color or turbidity of the sample does not interfere with the optical signal 1-3.
Here we report on a simple biosensor for urea where an ammonium ion sensitive membrane served as transducer. The cocktail (containing PVC, plasticizer, nonactin as ionophore and a pH sensitive dye) was spread onto a silylized glass substrate via spin coating. The transducting membrane was coated by a very thin polyurethane layer on which urease enzyme was immobilized by using glutaraldehyde as cross linking agent.
The sensor response to urea was determined by measuring the attenuation of a HeNe laser. The laser beam was coupled into and out of the glass plate using commercially available polyacetate gratings providing inexpensive sensor.
The useful concentration range of the waveguide based flow injection device was 0.05 - 3 mM urea with a response time of 15 seconds.
The financial support of the OTKA (T030968) and FKFP (0572/1999) are highly appreciated.
1 O.S. Wolfbeis: Trends Anal. Chem. (1996)15, 225-232
2 R.A. Potyrailo, S.E. Hobbs, G.M. Hieftje: Fresenius J. Anal. Chem. (1998) 362, 349-373.
EFFECT OF OXYGEN FREE RADICALS ON THE INTERNAL DYNAMICS OF ACTIN
Electron paramagnetic resonance technique (EPR) was used to study the effect of oxygen free radicals on different forms of actin. Reaction of -SH-containing compounds with Ce(IV)-ions in the presence of spin trap phenyl-N-tertier-butylnitrone (PBN) results in the appearance of nitroxide free radicals. The EPR spectrum is characteristic of a strongly immobilized spin label that is rigidly attached to the protein. It has been already reported that Ce(IV) complexed to nitrilotriacetic acid (NTA) oxidizes sulfhydryl compounds via thiyl free radicals, which can be trapped by PBN.In monomeric actin the most reactive -SH groups are Cys-235 and Cys-374, whereas in its polymerized form the most accesible thiol group is Cys-374. This latter group is not very far from the high affinity cation and nucleotide binding sites, therefore the protein conformation can be studied in both forms of actin in the presence of oxidizing agent.
The free radical concentration after Ce(IV)-NTA treatment was 0.42 mole of free radical/mole of protein in G-actin, whereas 0.37 mole of free radical/mole of actin was detected in F-form. The lineshape of the EPR spectrum was similar to the nitroxide EPR specrum indicating that these radicals are responsible for the EPR spectrum. The hyperfine splitttings obtained in this study were about the same for both forms of actin (G-actin: 2Azz=63.71 G, F-actin: 2Azz=63.54 G). It shows that the mild oxidation by Ce(IV)-NTA can modify the protein structure in the neighbourhood of the thiol sites.
Selective blocking the Cys-374 thiol group with N-etyl-maleimide before Ce(IV)-NTA treatment reduced markedly the concentration of the attached free radicals. The double integral of the EPR spectrum showed that 65 % of spin traps are localized on the Cys-374 residue. The thiol groups of proteins are known to protect biological tissues against oxidative stress and it has been thought that thiyl radicals plays a critical role in this process.
(This work was supported by grants from the National Research Foundation
(OTKA T 030248, CO-123, CO-272), INCO COPERNICUS EU ERBIC 15CT 960821 and
Ministry of Health (T 06 017/96).)
GLOBAL AND LOCAL CONFORMATIONAL CHANGES IN MYOSIN INDUCED BY AMP.PNP
Hartvig, N.1*, Gaszner, B.1, Schäffer, B.2, Lõrinczy, D.2, Belágyi, J.1
1Central Research Laboratory, University Medical School and 2Institute of Biophysics, Pécs, Hungary
It was already published that an isothiocyanate-based spin label (TCSL) is more sensitive to the domain orientation in myosin head than the widely used maleimide spin label (MSL). The former has a particular orientation which reflects smaller changes and does sense the internal rearrangement of the segments (Belagyi et al., 1994; Eur. J. Biochem. 224: 215-222). We report now the effect of the nonhydrolyzable ATP analogue AMP.PNP on the dynamics and structural stability of myosin head using EPR spectroscopy and DSC technique.
Skeletal muscle fibres isolated from psoas muscle of rabbit were spin - labelled with isothiocyanate-based probe molecules at the reactive sulfhydryl site (Cys-707) of the catalytic domain of myosin. AMP.PNP increased the orientational disorder of myosin heads, a random population of spin labels was superimposed on the ADP-like spectrum evidencing conformational and motional changes in the internal structure of myosin heads. Saturation transfer EPR measurements reported increased rotational mobility of spin labels in the presence of AMP.PNP corresponding to weakly binding state of myosin to actin. Measurements on MSL-fibres in the presence of AMP.PNP showed that about half of the cross-bridges dissociated from actin (Fajer et al., 1988; Biophys. J. 53: 513-524). This fraction had dynamic disorder, whereas the other population had the same spectral feature as in rigor. The results suggest that in the presence of AMP.PNP the attached heads have the same global orientation as in rigor, but the internal structure undergoes a local conformational change.
The thermal unfolding is a complex process characterizing three discrete domain regions with different thermal stability. The unfolding at 52.2 °C characterizes the catalytic domain of myosin, whereas the transition at Tm=58.8 °C represents the unfolding of the rod-like region. The last transition (Tm=67.8 °C) refers to the nucleotide binding to myosin.
This work was supported by grants from the National Research Foundation
(OTKA T 030248, CO-123, CO-272), INCO COPERNICUS EU ERBIC 15CT 960821 and
Ministry of Health (T 06 017/96).
EPR STUDY ON CHROMIUM-SENSITIVE AND RESISTANT MUTANTS OF SCHIZOSACCHAROMYCES POMBE
Lysine and leucine auxotrophic, heterothallic (h+,h-) strains of Schizosaccharomyces pombe were used to study chromium(VI)-sensitive and resistant mutants. The effect of Cr(VI) anions on plasma membrane was detected in vivo by spin label electron paramagnetic resonance (EPR) spectroscopy. 5-doxyl stearic acid (5-SASL) and 3-doxyl butyric acid (HO-185) spin probes were incorporated into the membrane, and the order parameter S was derived from the nitroxide EPR spectra at different temperatures (0-25 oC) to calculate the rotational dynamics of the membranes components. Mutants in control experiments exhibited altered structural transitions both in 5-SASL- and HO-185-membranes in comparison with their parental strain suggesting alterations in membrane composition of these mutants. Addition of 225 M K2Cr2O7 significantly effected the phase transition temperatures of the 5-SASL-membrane of wild-type strain CRW-6 and sensitive mutant CRS-6.51, but chromium ions slightly increased the phase transition temperature for resistant mutant CRR-6.66 as revealed by the HO-185 label.
DETERMINATION OF IONIC SURFACTANTS BY SOLID STATE POTENTIOMETRIC SENSOR ARRAY
Balázs Csóka*, Barna Kovács and Géza Nagy
Research Group for Chemical Sensors, MTA-JPTE, Department of General and Physical Chemistry, Janus Pannonius University, H-7624 Pécs, Ifjúság u. 6, Hungary
Ionic surfactants can be measured by colorimetric two-phase titration or by potentiometric titration using surfactant sensitive indicator electrodes1. In this work we describe all solid state potentiometric surfactant sensitive electrodes.
Teflonized graphite rods were used as support. They were covered at first with polypyrrole film by electropolymerization. Then they were coated by plasticized PVC membrane containing different sensing materials. These sensing materials were made of ion-pairs of different cationic and anionic detergents in 1:1 molar ratio. Three ion-pairs (CTA-ODS, DDMA-TPB, TOA-LS)3 were synthesized by simple mixing the aqueous solutions of the components in stochiometric ratio1,2. The three electrodes were placed into a flow cell and characterized by using a flow injection apparatus using SCE as reference electrode.
Anionic surfactants (C8 - C12) could be determined in the range of 5M - 1mM. The detection limit strongly depends on the carbon chain length of the analyte and on the sensing material used.
The financial support of the OTKA (T030968) is highly appreciated.
1 Lj. Zelenka, M. Sak.-Bosnar, N. Marek, B. Kovács: Anal. Lett., (1989) 22, 2791-2802.
2 G.J. Mohr, T. Werner, I. Ohme, C. Preininger, I. Klimant, B. Kovács, O.S. Wolfbeis: Adv. Mater., (1997) 9, 1108-1113.
3 Abbreviations: Cetyltrimethylammonium ion (CTA), didodecyldimethylammonium ion (DDMA), tetraoctylammonium ion (TOA), octadecylsulphonate ion (ODS), tetraphenylborate ion (TPB), dodecylsulphate ion (LS).